Vesiculobullous disorders
Vesiculobullous (blistering) eruptions are seen in many systemic illnesses, e.g. exanthematous fevers, e.g. varicella or primary skin disorders, e.g. infections, hypersensitivity reactions, autoimmune or hereditary disorders, etc.
(Table 25.8).Differential diagnosis depends on the age, morphology/ distribution of lesions, precipitating factors and presence of coexisting mucosal/systemic involvement. Family history indicates hereditary disorders, e.g. epidermolysis bullosa.
Skin biopsy to determine the depth of blisters is an extremely valuable in diagnosis of vesiculo-bullous disorders, which may be broadly divided into following categories:
a. Intracorneal or subcorneal blisters, e.g. miliaria, candidiasis, are very tiny, fragile and easily missed. Some cases present only as desquamated skin.
b. Intra-epidermal blisters, e.g. infections, pemphigus, etc. are usually flaccid, easily ruptured and heal without scarring. Niklosky sign, i.e. lateral extension of lesion on pressure is positive in some cases.
TABLE 25.8: Causes of vesico-bullous eruptions
• Infections
Bacterial: Impetigo, staphylococcal scalded skin syndrome Viral: Chickenpox, HSV, VCZ, hand-foot-mouth syndrome Others: Inf. scabies, Hookworm*, Rickettsial pox
• Hypersensitivity
Erythema multiforme
Steven-Johnson syndrome
Toxic epidermal necrolysis
• Autoimmune
Pemphigus/pemphigoid
Dermatitis herpeteformis
• Hereditary
Epidermolysis bullosa
Porphyria tarda
Incontinentia pigmenti
• Extrinsic factors
Burns, insect bites
Miliaria
*at the site of skin entry.
HSV: Herpes simplex; VCZ: Varicella zoster
c. Sub-epidermal blisters, e.g. Steven-Johnson syndrome and its variants, are tense, thick-walled and more durable. Some important Vesiculobullous disorders with primary skin involvement are as follows:
Staphylococcal scalded skin syndrome (SSSS, Ritter syndrome), is almost always seen in infants or children below 5 years of age, presenting with a spectrum of clinical manifestations ranging from localized bullous impetigo to generalized skin lesions with systemic involvement.
Etiologically, SSSS is caused by epidermolytic or exfoliative exotoxins of phage group 2 staphylococci (strain 71, 55), usually infecting nasopharynx, umbilicus or superficial abrasion.
Clinically, a typical case presents with:
a. Prodromal phase with fever, malaise, irritability and extreme tenderness all over the skin for few hours;
b. Exfoliative phase, beginning as a generalized macular or erythematous rash, specially in flexural or periorificial region that rapidly evolves to form ill- defined bullae/blisters filled with clear fluid. Skin surrounding the bullae separates easily by gentle finger pressure (Nikolsky sign). Peeling of large sheets of epidermis leaves behind denuded, moist areas (Fig. 25.11), with complications, e.g. secondary infection and fluid-electrolyte imbalance.
Circumoral erythema and radial crusting/fissuring around eyes, nose and mouth is characteristic in SSSS, though intra-oral lesions are absent. Pharyngitis and conjunctivitis is common as also systemic infections, e.g. septicemia or pneumonia.
c. Desquamative phase begins after 2-5 days, healing the ulcers without scarring.
Atypical cases may present with localized bullous impetigo or generalized erythema without blister formations. Diagnosis rests on typical evolution and distribution of lesions with positive Nikolsky sign, confirmed on skin biopsy to show intra-epidermal plane of cleavage and characteristic absence of inflammatory infiltrate. Intact bullae/blisters are essentially sterile, though cultures from suspected sites, e.g. throat may reveal source of toxin production.
Treatment of these cases includes:
a. Systemic antibiotic therapy with IV/PO penicillinaseresistant penicillin, e.g. cloxacillin or amoxicillin/ clavulanic acid.
b. Local care: Gentle moistening and cleaning of skin with normal saline followed by application of bland emollient ointment (topical antibiotics are useless, unless secondary infection is present).
c. Treatment of complications, e.g.
fluid and electrolyte imbalance or systemic infection, e.g. pneumonia, septicemia, etc.Steven-Johnson syndrome (SJS) is the commonest and prototype manifestation of acute vesicobullous hypersensitivity reactions to various infections, drugs or other agents (Table 25.9) in young children below 10 years of age.
Other uncommon presentations include Erythema multiforme and Toxic epidermal necrolysis, discussed later, which share a common pathogenesis but with variable severity and age of presentation.t
Clinically, SJS is often preceded by mild viral respiratory illness and characterized by rapidly progressive involvement of skin and at least two mucus membranes, (Fig. 25.12) as follows:
a. Generalized skin lesions, which begin as tender, dusky, erythematous macules and progress rapidly with central necrosis to form vesicles, bullae and large denudated areas over face, trunk and extremities. New lesions appear in crops for next 1-2 weeks.
b. Mucosal involvement of at least two of the following sites: (a) eyes with conjunctivitis or keratitis/corneal ulcers, (b) oropharynx with edema, erythema, ulceration, crusting and burning sensation, (c) anogenital mucosal lesions, and (d) internal mucosal lesions manifesting as cough, diarrhea/dysentery or hematuria.
c. Secondary complications in acute phase include:
(a) dehydration due to insensible fluid loss from
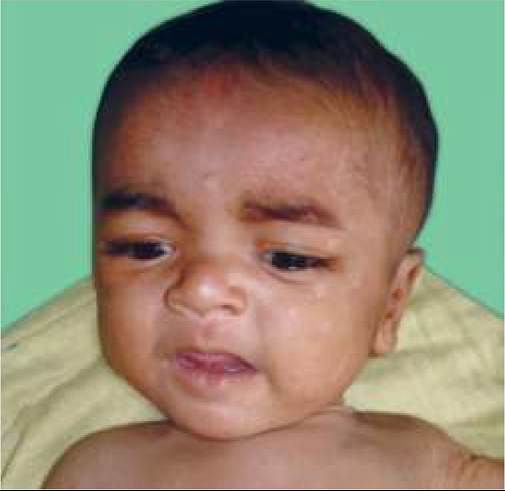
Fig. 25.11: Staphylococcal scalded skin syndrome.
TABLE 25.9: Causes of Steven-Johnson syndrome
• Infections:
- Viral: HSV, EBV, HBV, enteroviruses
- Bacterial: Streptococci gr A, tuberculosis
- Mycoplasma pneumoniae
• Drugs:
- Anticonvulsants: Phenytoin, Phenobarb, Carbamazepine
- Antibiotics: Sulfonamides*, penicillin, isoniazid
- Others: NSAIDs, captopril, radiotherapy
• Neoplasia: Leukemia, lymphoma
• Miscellaneous: Photosensitivity, pregnancy
*commonest cause.
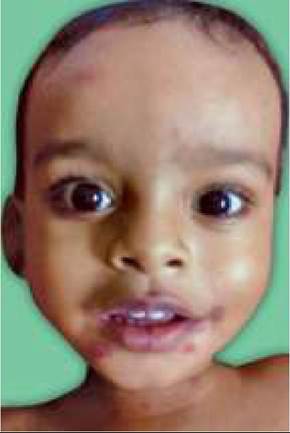
Fig.
25.12: Steven-Johnson Syndrome: (A) Mucosal Lesions; (B) Skin Lesions.(Courtesy: Dr Uday Khopkar)
lesions, (b) superadded local infection or septicemia, (c) satellite infections, e.g. bronchopneumonia, arthritis, panophthalmitis, myocarditis, hepatitis, etc.
d. Self-limiting course with complete healing in 4-6 weeks, though rare late complications include: (a) corneal scarring and visual impairment, and (b) bronchial/ esophageal/anal/vaginal strictures.
Diagnosis is clinical, supported by history of drug exposure or preceding viral illness. Skin biopsy reveals sub-epidermal plane of cleavage.
Management is largely supportive, including:
• IV Fluid and electrolyte correction in acute stage.
• Nutritional support with IV fluids or oral feeds. Topical anesthetics, e.g. lidocaine jelly may be used before feeds to facilitate oral feeding.
• Systemic antibiotics for secondary infections.
• Cleaning of denuded areas with saline/Burrow's solution compresses and paraffin/hydrogel dressings.
• Eye-care with antibiotic or moistening eye drops.
• Steroids are not routinely recommended though a short course of high-dose steroids may be useful in severe disease with deeper mucosal lesions.
Erythema multiforme (EM), is more common hypersensitivity reaction in older children gt;10 years. HSV infection has been implicated in 60% cases of EM and nearly all cases of recurrent EM.
Clinically, primary lesions in EM are described as target (bulls-eye) lesions, i.e. papules with erythematous margins, inner pale ring and central necrosis. Unlike SJS, EM is distinguished by:
• Lesions largely limited over extensor aspects of upper limbs, usually sparing face and trunk.
• Rarity of mucosal involvement except buccal mucosa. However, ~25% cases may present only with mucosal lesions without skin lesions.
• Absence of prodromal symptoms.
• Shorter course with complete resolution in two weeks, without sequelae.
Treatment is supportive as for SJS, though prophylactic acyclovir therapy for 6 months may be beneficial in cases of recurrent EM following HSV infection.
Toxic epidermal necrolysis (TEN) or Lyell syndrome, is most severe and potentially fatal hypersensitivity reaction, though relatively uncommon in childhood.
Clinically, it is characterized by:
• Widespread blister formation, followed by full thickness skin loss in sheets and positive Nikolsky sign. However, mucosal lesions are less severe.
• Marked constitutional toxicity with fulminant progressive course and frequent complications, e.g. shock and septicemia.
• Absence of target lesions (d/d erythema multiforme) Diagnosis is confirmed on skin biopsy that reveals full thickness epidermal necrosis. D/D mainly involves SSSS, which is common in infants and lesions often spare mucosa, palm and soles.
Management of TEN needs to be more intensive, as for the burns or SJS. Systemic steroids are of no use.
Pemphigus vulgaris is an autoimmune disorder with high IgG titers against an epidermal intercellular substance-a glycoprotein desmoglobin III. Although an acquired disorder, P. vulgaris may also be seen at birth in neonates born to mothers with pemphigus.
Clinically, it is characterized by:
• Large flaccid bullae on non-erythematous base (d/d SJS), with positive Nikolsky sign, mostly over pressure points, face, groin and axilla. Blisters rupture and enlarge peripherally leaving large denuded areas with slow healing.
• Waxing and waning course, often leading to malnutrition, debility and terminally, the death.
Diagnosis rests on skin biopsy showing intradermal cleavage and characteristic anti-desmoglobin IgG antibodies on immunofluorescent staining.
Treatment: High-dose systemic steroid therapy is the treatment of choice, though some cases need cytotoxic therapy with methotrexate, cyclophosphamide or azathioprine.
Other forms of pemphigus are rare in children.
Chronic bullous disease of childhood is a IgA mediated blistering disorder, common in children lt;5 years with itchy, tense bullae of erythematous lesions, usually grouped around orifices, e.g.
perioral, perinasal, perianal regions or in lower abdomen and buttocks. New lesions appear around previous lesions (String of pearl appearance). Disease is self-limiting after 2 years. Mild cases may be treated with dapsone, while severe cases required oral steroids.Dermatitis herpetiformis, most commonly seen in 2-7 years age group, is characterized by:
a. Symmetric, grouped, intensely pruritic papules or vesicles (pleomorphic) sparing mucus membranes, mainly over knees, elbows, shoulders and buttocks. Lesions often mimic scabies, popular urticaria, insect bites or eczema.
b. Sub-epidermal blistering on skin biopsy.
c. Unknown etiology, though autoimmune pathology is suspected due to close association with Gluten sensitive enteropathy and HLA B-8 in gt;75% cases.
Treatment includes oral sulfapyridine or dapsone (most effective), local anti-pruritic agents and gluten-free diet. Epidermolysis bullosa (EB) is group of congenital/ hereditary disorders with variable clinical severity and inheritance pattern, the common denominator being development of blisters after minor trauma that exacerbate during warm weather and heal with/without scarring.
Clinical spectrum spans from mild autosomal dominant disease (EB simplex) without mucosal involvement or scarring to potentially fatal (Junctional EB) or severely deforming (dystrophic EB) variants of autosomal recessive origin. Skin biopsy reveals sub-epidermal blistering.
Epidermolysis bullosa simplex, the commonest variant, usually manifests at birth or soon after, with recurrent development of blisters over scalp and dorsal extremities, which heal without scarring. Mucosal involvement is rare. Blisters may be precipitated by minor trauma or warm weather. Secondary infections are common. Propensity to blister decreases with age.
Therapy is largely supportive including-avoidance of trauma, local care, topical antibiotics and management of systemic complications. Although blisters should be drained by puncturing, the top should be left intact to protect underlying skin.
Incontinentia pigmenti is a rare X-linked dominant disorder, which may be fatal in males. on flexor aspects of legs/arms, is uncommon in childhood but may overlap second stage.
Other ectodermal lesions, e.g. alopecia or dental anomalies are seen in ~80% cases, while neurological manifestations, e.g. mental retardation, seizures, etc. and/or ocular lesions, e.g. cataracts, optic atrophy, etc. are present in ~30%.
Diagnosis is largely clinical, supported by wood-lamp examination for pigmentary abnormalities. Skin biopsy of blistering lesion reveals intra-epidermal cleavage.
Management includes treatment of non-cutaneous abnormalities (skin lesions are benign) and genetic counselling.
25.7