Genomic Comparison of MAP Strains
The advancements in DNA sequencing technologies, particularly next-generation sequencing and development of diverse new computational tools for comparative genomics, have expedited MAP genome research in the past decade or so.
The first complete annotated genome sequence of a MAP-C strain (K10) was published in 2005 (Li et al., 2005), subsequently revised following optical mapping (Wu et al., 2009) and resequenced (Wynne et al., 2010). The first complete genome sequence of a MAP-S strain (Telford 9.2) was recently reported (Brauning etal., 2019), although draft sequences of MAP-S strains S39 7 and JIII-386 were published by Bannantine et al. (2012) and Mobius et al. (2015), respectively.‘Corresponding author: karen.stevenson@moredun.ac.uk
© CAB International 2020. Paratuberculosis: Organism, Disease, Control, 2nd Edition
(eds M.A. Behr et al.)
These genome sequences provide reference genomes for mapping unassembled sequence reads of other isolates and for comparative genomics that can quickly identify large sequence polymorphisms (LSPs) including insertions, deletions, inversions, translocations and duplications, and individual SNPs that contribute to the unique genetic profiles of MAP strains. Other draft genome sequences are available in public databases as well as raw sequence data for a large number of MAP isolates, and this resource will expand as WGS becomes cheaper over time. The salient features of the MAP genome were described in the previous edition of this book (Paustian et al., 2010), this chapter will focus on comparative genomics between MAP strains and genomic diversity of MAP isolates.
6.2.1 Phylogenetic relationships among MAP strains
The first comprehensive WGS study to clarify the phylogenetic relationships between previously designated strains was published by Bryant et al. (2016) using a global panel of 141 MAP isolates (modified in Fig.
6.1) that included the two major lineages corresponding to the ‘Sheep-type’ or ‘Type S’ and the ‘Cattle-type’ or ‘Type C’ designated by Collins et al. (1990). Type C is synonymous with the Type II strains, and Type S with the Type I/III strains, as defined by pulsed-field gel electrophoresis (Stevenson et al., 2002). Type S and Type C strains differ with respect to the ease with which they can be isolated on artificial media and their respective growth rates (see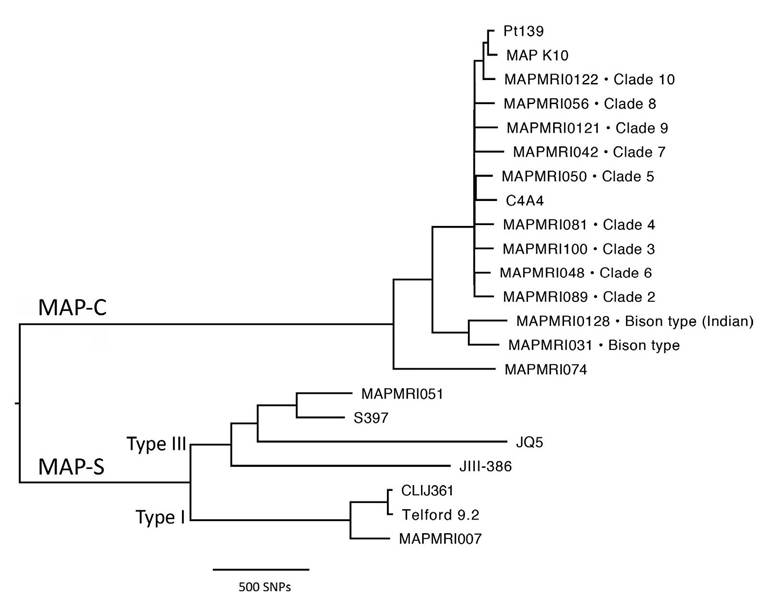
Fig. 6.1. Midpoint rooted whole genome single nucleotide polymorphism (SNP)-based phylogenetic tree of a global panel of Mycobacterium avium subsp. paratuberculosis (MAP) strains. Tips are annotated with strain names and, when applicable, MAP-C clade IDs as defined by Leao et al., 2016. MAP-S and C lineages are labelled, as well as Type I and III MAP-S sub-lineages. Publicly available fastq files or assembled contigs were reference mapped to the MAP K10 genome and SNPs were identified using methods described by Bryant et al., 2016.
Chapter 18, this volume) and also in terms of their virulence and pathogenicity (see Sections
6.2.3 and 6.4). However, since the original designation of ‘Sheep-type’ and ‘Cattle-type’ strains, it has become apparent that strain type is not always linked to host species provenance (see Section 6.3.1) and consequently this nomenclature can be confusing. Therefore, we propose that the two lineages are designated MAP-S and MAP-C and any reference to host species is avoided. These terms will be used throughout this chapter.
MAP-S is sub-divided into two sub-lineages. One sub-lineage comprises the ‘Intermediate’ strains (Collins et al., 1990) or ‘Type III’ strains described by de Juan et al. (2005), which were hypothesized to be intermediate between Type S and Type C, a supposition subsequently not supported by phylogenomics (Fig. 6.1; Alexander et al., 2009; Mobius et al., 2015; Bryant et al., 2016).
This sub-lineage also includes the sequenced Arabian camel isolates (Ghosh et al., 2012; Bryant et al., 2016). The other sub-lineage corresponds to the Type I pigmented ovine isolates from the UK described by Stevenson et al. (2002). Pigmentation was originally thought to be a unique characteristic of Type I strains but a few pigmented isolates have since been typed as Type III (Biet et al., 2012) and a nonpigmented isolate as Type I (Bryant et al., 2016). Furthermore, the draft genome of a MAP-C pigmented isolate, MAP C4A4, has been published (Barbosa et al., 2017). No genetic polymorphisms or the presence or absence of any particular gene that could be unique to pigmented MAP isolates have been identified by genomic comparisons to date.MAP-C also has a sub-lineage that encompasses the group of isolates designated as ‘Bison type’ or ‘Type B’. This strain type was first described by Whittington et al. (2001a) and was defined by the number of copies of C or T at base pair position 223 in the insertion sequence IS1311, which could differentiate between isolates from US bison (Bison bison) and other cattle isolates. More recently, a new genotype designated ‘Indian Bison type’ has been described, differentiated from other Type MAP-C strains by deletion of base pairs at positions 64 and 65 in IS1311 (Sohal et al., 2013). A divergent MAP-C isolate from the Netherlands (MAPMRI074) was included in the global panel of isolates sequenced by Bryant et al. (2016), but has not been further characterized to date. This suggests that there may be further undiscovered genetic diversity even within the MAP-C lineage for which a far greater number of isolates have been sequenced.
6.2.2 Genotypic differences among MAP strains
The two major MAP lineages can be distinguished by genetic polymorphisms, in particular by the presence/absence of LSPs, which can be the result of either insertions or deletions. These have been determined by microarray comparisons and in silico analysis of WGS data from MAP-C and S isolates and Mycobacterium avium subsp.
hominissuis strain 104. The evolution and phylogeny of MAP from M. avium is covered elsewhere (see Chapter 5, this volume).MAP-S strains are characterized by the presence of four clusters of open reading frames (ORFs) that are absent from MAP K-10 and most of which are present in the M. avium hominis- suis genome. These clusters have been consistently detected in all MAP-S strains analysed to date (Semret et al., 2006; Paustian et al., 2008; Alexander et al., 2009; Castellanos et al., 2009; Bannantine et al., 2012; Mobius et al., 2015) and details are given in Table 6.1. Comparative genomics has revealed other polymorphisms absent from MAP-C strains that may reflect genetic diversity within MAP-S strains but alternatively could be due to sequencing errors or discrepancies between the methodologies used (Alexander et al., 2009; Castellanos et al., 2009; Mobius et al., 2015). In the converse direction, MAP-C strains are differentiated from MAP-S strains by the presence of two clusters of ORFs that are consistently absent from MAP-S strains (Table 6.2).
Mobius et al. (2015) report fewer than 200 SNPs among three MAP-C strains compared with about 1000 among three MAP-S strains, consistent with a higher heterogeneity within the MAP-S lineage (in part due to the large number of SNP differences between Type I and III strains) and high similarity among MAP-C strains.
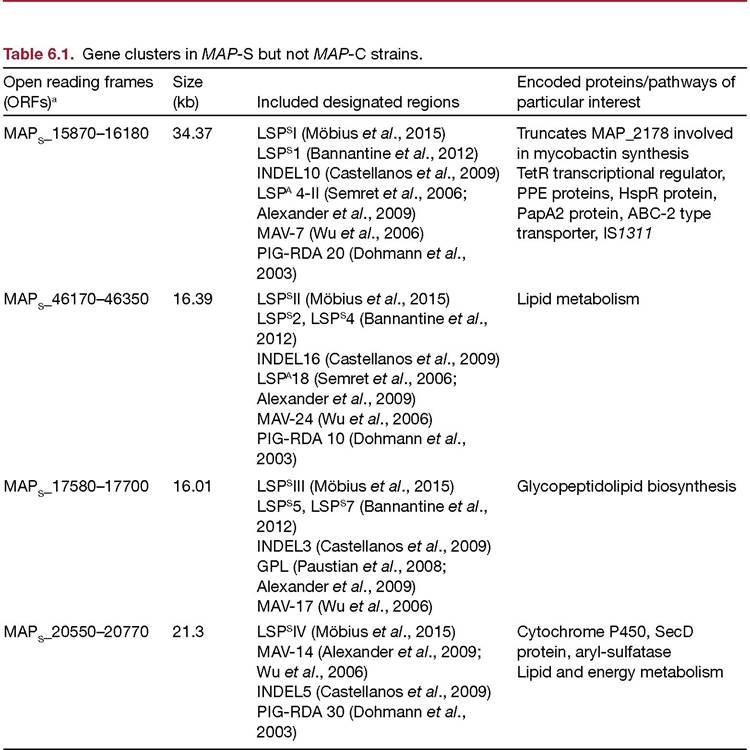
aORFs as designated by Bannantine etal., 2012.
Table 6.2. Gene clusters in MAP-C but not in MAP-S strains.
Open reading frames (ORFs) Included designated regions
MAP_1491 to MAP_1484 c LSPa20 (Alexander et al., 2009)
INDEL6 (Castellanos et al., 2009)
MAP_1728 c to MAP_1744 INDEL7 (Castellanos et al., 2009)
Del-2 (Marsh et al., 2006; Alexander et al., 2009)
MAV-14 (Wu et al., 2006)
RDA3 (Marsh and Whittington, 2005)
Genes/pathway of particular interest
Pyruvate dehydrogenase complex
yfnB (predicted hydrolase)a mmpS and mmpL genesab MAP_1729 c & MAP-1730 (hypothetical proteins)a fabG3 (lipid biosynthesis) acg, devSb
aTranscription upregulated in monocyte-derived macrophages (MDMs) in study by Zhu et al.
(2008). bAlso identified by representational difference analysis (Marsh and Whittington, 2005).6.2.3 Insights into the virulence and pathogenicity of MAP strains
Comparative genomics provides insights into the virulence and pathogenicity of MAP strains. Analysis of strain-specific polymorphisms reveals presence/absence of genes that may influence virulence and pathogenic traits as detailed in Table 6.3. Many of the lineagespecific polymorphisms encode hypothetical proteins of unknown function. Targeted studies will now be required to determine the functional impact of these natural polymorphisms between strains. Additionally, comparative genomics of genetically similar strains can provide insights into virulence and pathogenicity. A study of MAP-C vaccine strains by Bull et al. (2013) identified genomic variations associated with attenuation that were probably derived in a classical manner by selective subculture on different media. Twenty-five MAP-specific gene deletions were identified of which at least one (MAP3 730; S-adenosylmethionine- dependent methyltransferase with homologues to tellurite resistance genes) could be linked to a phenotypic change that would decrease its virulence and persistence in the host. Timms et al. (2015) performed a comparison of MAP-C strains isolated from humans and animals, which suggested that although the genomes are closely related, there may be key differences in virulence factors, including the PE/PPE (proline-glutamate/proline-proline- glutamate motif) genes, mammalian cell entry (mce) operons and the mycobactin cluster.
6.3
More on the topic Genomic Comparison of MAP Strains:
- Comparison of the Virulence and Pathogenicity of MAP Strains
- Genomic Epidemiology of MAP
- Cultural Requirements of Different Strains of MAP
- Comparative Genomics and Genomic Epidemiology of Mycobacterium avium subsp. paratuberculosis Strains
- COMMON INBRED STRAINS
- Comparison of Culture Methods
- GENOMIC AND RAPID RESPONSE PATHWAY TO VITAMIN D
- GENOMIC CONSIDERATIONS FOR THE PATHOLOGIST
- DISTINCT HIV-1 STRAINS AND NEUROLOGICAL DISEASE
- Genetic Diversity of Mycobacterium bovis Strains in Tanzania
- A CROSS-CULTURAL COMPARISON OF SECTORAL LINKS: MALACCA
- GENOMIC BASIS OF CD8-MEDIATED PROTECTION: CLUES FROM THE WHOLE HUMAN GENOME MICROARRAY STUDIES