AUTOLOGOUS CELL-MEDIATED KILLING VIA INTERCELLULAR CONTACTS
Monocyte/Macrophage-Mediated T Cell Apoptosis
CD4+ T Cell Apoptosis
In the lymph nodes of HIV-seropositive individuals, HIV-mediated immune activation is observed,1-2 and apoptosis occurs predominantly in the bystander uninfected cell populations, especially in the presence of APCs, such as M0.3-13 The key role of HIV-infected M0 in causing CD4+ T cell depletion was first suggested by results obtained from the severe combined immunodeficient (SCID)-HIV mouse model.14 It was observed that monocytotropic viruses resulted in enhanced and accelerated apoptosis of the repopulated CD4+ T cell population.14 Also, monkeys infected with HIV-1, which does not infect simian monocytes∕M0, show no signs of disease, have stable CD4+ T cell counts, and do not show accelerated T cell apoptosis.15,16 These observations suggested that M0 might play a crucial role in CD4+ T cell depletion after HIV infection.
CD4 cross-linking by the HIV envelope glycoprotein (gp)120 was shown to prime T cells for activation-induced apoptosis, via the upregulation of expression of Fas and Fas ligand (FasL) on uninfected T cells and M0, respectively (Figure 15.1).3,17 Macrophages express basal levels of FasL that are significantly upregulated after infection with HIV,3 and FasL expression is enhanced compared with monocytes from HIV-negative controls.18 HIV-infected M0 (and, to a lesser extent, uninfected M0) were shown to kill Fas-sensitive T cell targets3 in a major histocompatibility complex-unrestricted and Fas/tumor necrosis factor (TNF)-dependent manner.4 FasL is elevated in peripheral blood mononuclear cells (PBMCs, which contain monocytes)19-21 from HIV-infected patients.
The plasma level of soluble FasL is increased in HIV-positive patients, correlates with HIV RNA burden, and can be used as a predictive marker for the progression to acquired immunodeficiency syndrome (AIDS).22,23 A marked increase in M0-associated FasL was also detected in lymphoid tissue from HIV-positive subjects, which correlates with the level of tissue apoptosis.24 Dexamethasone inhibits CD4+ T cell deletion in a dose-dependent manner. The deletion of normal CD4+ T cells by M0 from HIV-infected patients was also inhibited by dexamethasone. Furthermore, upregulation of Fas expression on T cells exposed to anti-CD4 and gp120∕IgG, which predisposes T cells to Fas-mediated apoptosis, is inhibited by dexamethasone in a dose-dependent fashion. Dexamethasone inhibits the M0-mediated deletion of CD4+ T lymphocytes in HIV-infected per- sons.25 M0-mediated CD4+ T cell apoptosis has implications in vivo, because levels of tissue apoptosis directly correlate with levels of M0-associated FasL.24 Thus, FasL may be one of the main mediators of uninfected CD4+ T cell death by monocytes/M0.As well as the involvement of Fas-FasL interactions, TNF-α was reported to trigger apoptosis in uninfected CD4+ T cells upon HIV infection.4 HIV-infected M0-mediated killing of uninfected CD4+ T cell blasts can be partially reduced by the administration of soluble TNF-receptor (TNFR) decoys,26 and TNF may contribute to apoptosis induced by gp120-mediated cross-linking of CD4.27
Although FasL and, to a lesser extent, TNF seem to be the mediators of M0-mediated apoptosis of CD4+ T cells, antigen-induced apoptosis of CD4+ T cells from HIV-infected individuals can be mediated by other members of the TNFR family known to induce apoptosis, such as the TNF-related apoptosis-inducing ligand (TRAIL∕APO2L).28 T cells from HIV-positive individuals are susceptible to TRAIL-mediated killing, in contrast to cells from control donors, and activation-induced cell death (AICD) is partially inhibited by antagonistic TRAIL-specific antibodies.29 The fact that AICD in patients with HIV infection may be partially inhibited using antagonistic TRAIL∕APO2L-specific antibodies28 suggests that TRAIL∕APO2L and TRAIL∕APO2L receptor dysfunction may contribute to HIV pathogenesis.
After infection with HIV, TRAIL is released by M0, and exogenous HIV- encoded Tat protein upregulates the production of TRAIL by primary M0 in vitro,30 indicating that a TRAIL-dependent mechanism of destruction of bystander CD4+ T cells may occur in vivo, which might be triggered by Tat produced by HIV-infected cells. Tat induces TRAIL in PBMCs, and the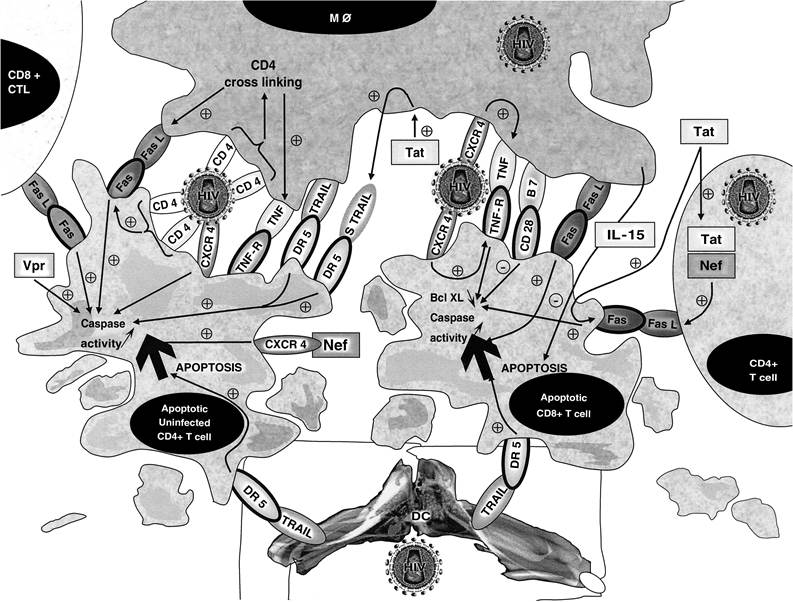
FIGURE 15.1 Intercellular contacts and soluble factors involved in autologous cell-mediated killing during HIV infection. Uninfected CD4+ T cells in contact with M0 undergo apoptosis following CD4 cross-linking and the resultant upregulation of expression of Fas and FasL on the surface of CD4+ T cells and M0, respectively. Fas-FasL interaction triggers the apoptosis of uninfected CD4+ T cells via activation of the caspase pathway. CD8+ T cells in contact with M0 undergo apoptosis after CXCR4 stimulation by X4 viruses. TNF-TNFR2 interaction triggers apoptosis of CD8+ T cells via downregulation of antiapoptotic proteins of the Bcl-2 family, such as Bcl-xL, and via caspase activation. As CD28 stimulation increases the levels of antiapoptotic members of the Bcl-2 family, such as Bcl-xL, B7-CD28 interaction might rescue CD8+ T cells from M0-mediated CD8+ T cell apoptosis. DC could kill both CD4+ T cells and CD8+ T cells via TRAIL- DR5-mediated apoptosis.
majority of TRAIL is produced by the monocyte subset of PBMCs. A slight increase in cell surface expression of TRAIL was observed on monocytes, and sufficient TRAIL was secreted to be toxic for T cells. Remarkably, uninfected T cells are more susceptible to TRAIL than are HIV-infected T cells. The production of TRAIL by Tat-stimulated monocytes provides a mechanism by which HIV infection can destroy uninfected bystander cells.31
HIV-1-infected antigen-presenting monocytes are able to prime in vitro non-HIV-infected antigen-specific CD4+ T cells or peripheral blood CD4+ T cells to undergo apoptosis after antigenspecific restimulation.
Priming for apoptosis required two concomitant signals present on the same APC, an antigenic stimulus and a second signal provided by the HIV gp120 protein.6Because HIV gp120 binds to both CD4 and the chemokine receptors CXCR4 and CCR5 on the cell surface, it was suggested that the binding of gp120 not only to CD4 but also to the chemokine receptors can trigger apoptosis of uninfected T cells (Figure 15.2). Monoclonal blocking antibodies directed against the gp120 binding site of CD4 and against CXCR4 prevent the apoptotic signal
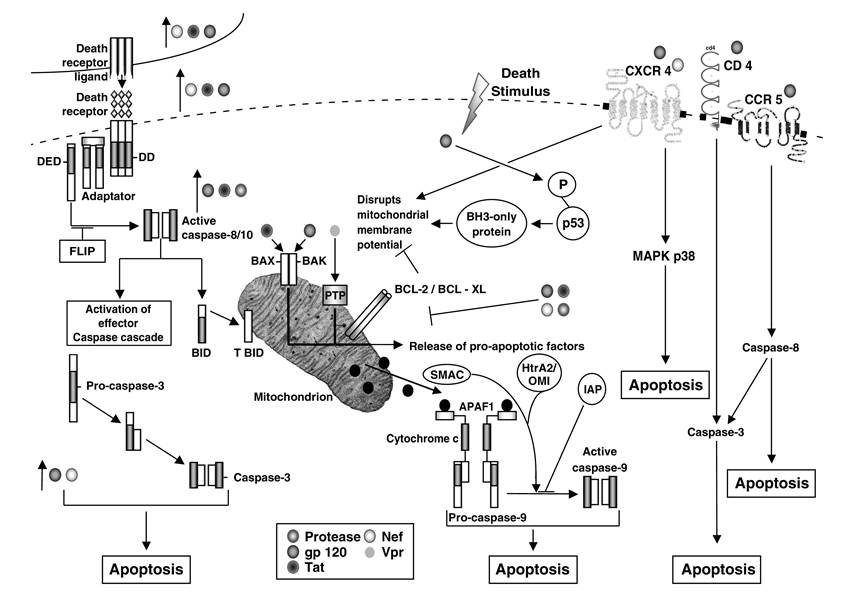
FIGURE 15.2 Molecular mechanisms involved in autologous cell-mediated killing during HIV infection. The extrinsic pathway is initiated by the binding of TNF-family death-receptor ligands to their cognate receptors. Through their death domains (DDs), multimerized receptors interact with the DDs adaptor proteins, which also contain death-effector domains (DEDs) that facilitate binding to procaspase-8 and procaspase-10 to form the death-inducing signal complex (DISC). As part of the DISC, the procaspases are cleaved into their active forms and initiate the intrinsic pathway of apoptosis. BID (BH3-interacting domain death agonist) is then cleaved to produce truncated (T)BID, and the effector caspase cascade is activated. Death-receptor-induced apoptosis can be blocked by FLIP (FLICE-like inhibitory protein), which inhibits the proteolytic processing of caspase-8. HIV-encoded proteins gp120, Nef, and Tat upregulate the expression of Fas and FasL. In addition, Tat upregulates the expression of TRAIL; gp120, Tat, and protease upregulate caspase-8; and gp120 and Nef increase the activity of caspase-3. The intrinsic apoptotic pathway is initiated by internal sensors, such as p53, that activate BH3-containing proteins and mediate the assembly of the proapoptotic members of the Bcl-2 family, including BAX and BAK, into hetero-oligomeric pores in the mitochondrial membrane.
This results in the release of proapoptotic factors—cytochrome c, SMAC (second mitochondria-derived activator of caspases), and HtrA2/OMI—into the cytoplasm. This is associated with the loss of mitochondrial potential, which can be blocked by antiapoptotic proteins Bcl-2/Bcl-xL. Release of cytochrome c promotes the formation of the apoptosome, which includes Apaf-1 (apoptotic protease activating factor 1) and procaspase-9. Autolytic activation of caspase-9 initiates the effector caspase cascade. Caspase activation is negatively regulated by IAPs (inhibitors of apoptosis proteins), which are counterbalanced by SMAC and HtrA2/OMI. HIV-encoded proteins also trigger the mitochondrial pathway: gp120 induces the phosphorylation of p53; Tat and gp120 promote BAX insertion into the mitochondrial membrane and subsequent release of cytochrome c; Vpr has a direct effect on the mitochondrial permeability transition pore (PTP); gp120, Tat, and Nef inhibit expression of Bcl-2, whereas protease cleaves it; and Nef inhibits the expression of Bcl-xL. CXCR4 activation by gp120 and Nef results in apoptosis via disruption of mitochondrial membrane potential and MAPK p38 activation. Gp120-mediated stimulation of CCR5 and CD4 triggers apoptosis via activation of caspase-3 and -8. in a dose-dependent manner.32 Using human peripheral blood lymphocytes, malignant T cells, and CD4 or CXCR4 transfectants, apoptosis was found to be induced by both cell surface receptors, CD4 and CXCR4.32 A role for CXCR4 in envelope-mediated CD4+ T cell apoptosis is further confirmed by the fact that a small molecule directed against CXCR4, AMD3100, inhibits cellsurface-expressed HIV-1 envelope-induced apoptosis.33 Also, HIV-1 envelope-mediated CD4+ T cell apoptosis is inhibited by the cognate ligand of CXCR4, stromal-derived Iactor-Ia (SDF-1α), again suggesting the involvement of CXCR4 in this phenomenon. The CXCR4-mediated CD4+ T cell apoptosis is Fas independent.32 In addition, HIV-1 virions, with mutant Env that binds CXCR4 but that are defective for CD4 binding or membrane fusion, induce apoptosis, whereas CXCR4- binding-defective mutants do not.33 Thus, HIV-1 virions can induce apoptosis through a CXCR4- or CCR5-dependent pathway that does not require Env∕CD4 signaling or membrane fusion. This suggests that HIV-1 variants with increased envelope/receptor affinity or co-receptor binding site exposure may promote T lymphocyte apoptosis in vivo by accelerating bystander cell death. Nevertheless, some reports suggest that HIV-1 envelope-mediated CD4+ T cell death depends mostly on envelope-CD4-receptor interactions, as CCR5-using, as well as CXCR4-using, envelopes elicited this response.34 These observations show that CD4+ T cell apoptosis can be mediated via CXCR4, and to a lesser extent CCR5, especially by HIV-1 envelope expressed on the surface of neighboring infected M0.32,35 The presence of CXCR4-mediated apoptosis, in addition to FasL-, TRAIL-, and TNF-mediated killing, indicates that CD4+ T cell apoptosis in HIV infection is a complex phenomenon that involves both immune activation and direct stimulation of CD4+ T cells by the HIV-1 envelope.CD8+ T Cell Apoptosis
As well as CD4+ T cell apoptosis, CD8+ T cell apoptosis is observed in peripheral blood isolated from HIV-infected subjects and could account for increased CD8+ T cell turnover during HIV infection. CD8+ T cells isolated from the peripheral blood of HIV-infected subjects are activated cells with an increased cell surface expression of Fas and TNFR2.13,28 Although Fas-FasL interaction was reported to be involved in CD8+ T cell apoptosis, the apoptosis seen in HIV infection is only partially inhibited by neutralizing anti-Fas and anti-FasL antibodies.13 The TNF-TNFR death pathway is also altered in HIV-positive individuals. Although an early report found that peripheral T cells from HIV-positive individuals were resistant to apoptosis that was induced by ligation of TNFR,36 more recent studies have shown that CD8+ T cells from HIV-positive individuals are susceptible to TNFR1- and TNFR2-induced apoptosis.13,37 The ligand for TNFRs, TNF, is detected at increased levels in the serum of symptomatic individuals, and elevated levels of soluble TNFR2 have been found to be predictive of HIV disease progression.38 TNF is partially responsible for activation-induced T cell apoptosis in animal models,39 and TNFR2 stimulation is involved in the apoptosis of mature CD8+ T cells.39-41 Apoptosis of CD8+ T cells during HIV infection has been shown to result from the interaction between membrane-bound TNF, which is expressed on the surface of activated M0, and TNFR2 expressed on the surface of activated CD8+ T cells.13 TNFR2 stimulation of T cells results in decreased intracellular levels of the apoptosis-protective protein Bcl-xL, a member of the Bcl-2 family.41 Impaired induction of Bcl-xL has also been observed in PBMCs isolated from HIV-infected patients. Thus, TNFR2 stimulation on CD8+ T cells by membrane-bound TNF, expressed on the surface of M0, decreases the intracellular levels of antiapoptotic proteins belonging to the Bcl-2 family and results in CD8+ T cell death. Susceptibility to TNFR- mediated apoptosis has been reported to be related to Bcl-2 expression, and early recruitment of caspase-3 and caspase-8 is needed to transduce the apoptotic signals. Thus, exacerbated TNFR- mediated cell death of T cells from HIV-infected individuals is associated with both alteration of Bcl-2 expression and activation of caspase-3 and caspase-8.37 Cross-linking of death receptors leads to the activation of an array of caspases, and both initiator caspase-8 and effector caspase-3 are activated after the ligation of TNFR1 and TNFR2 on T cells from HIV-positive individuals (Figure 15.2).37 The active forms of both caspases are expressed in vivo by several HIV-encoded proteins, such as Tat, Env, Nef, and Vpr.34,42-44 The function of CD8+ T cells killed by M0 via TNF-TNFR2 interaction is still under investigation, but TNFR2 stimulation has been shown to be involved in the depletion of cytotoxic T lymphocytes (CTLs).40 Also, CD8+ T cell apoptosis has been reported to be caused by APC, because removal of monocytes or addition of antibodies to CD80 and CD86 reduced apoptosis. A unique CD8brightCD28dim T cell population died after costimulation by monocytes. Because this population was increased in patients with undetectable viremia, abnormal APCs may contribute to continued CD8+ T cell exhaustion by inducing apop- tosis.18 It has also been reported that lymphocyte-reactive autoantibodies in HIV-1-infected persons facilitate the deletion of CD8+ T cells by M0.45
Dendritic Cell-Mediated T Cell Apoptosis
After measles and other viral infections and incubation with dsRNA, DCs become cytotoxic and, consequently, exhibit natural killer function through upregulation of type I interferon secretion that enhances TRAIL expression (Figure 15.1). In HIV infection, such a mechanism might be responsible for massive apoptosis of uninfected lymphocytes and increase specific immunity through cross-presentation of antigens from infected cells killed by DCs.46 Exposure of normal monocyte- derived DCs to HIV-1 virions leads to the induction of apoptosis in cocultured CD4+ as well as CD8+ T cells.47
Among several mechanisms of immune evasion used by HIV is the progressive destruction of virus-specific effectors, such as CD4+ T-helper cells, either through their direct infection during cognate interaction with infected DCs or through a bystander process that involves the upregulation of expression of death receptors and their ligands and the downregulation of expression of Bcl-2 family survival factors, leading to autocrine or paracrine destruction. After their recruitment into infected lymphoid sites, naive CD4+ T cell precursors might also be killed directly after specific priming and infection by HIV-infected DCs.48 Accordingly, during acute infection, rapidly proliferating HIV-specific memory CD4+ T cells are highly susceptible to HIV infection. Also, they have been found to contain higher levels of virus DNA than other memory CD4+ T cells,49 suggesting that they are preferentially infected in vivo and, consequently, that they are preferentially lost. Finally, destruction of activated HIV-specific CD4+ T cell effectors might result from fratricide mediated by FasL- or TNF-expressing killers that are generated by the persistent immune activation and the influence of HIV-encoded proteins. Failure to detect HIV-specific CD4+ T cells ex vivo might also be due to their anergy, resulting from interaction with peripheral blood DCs,50 their in vivo inhibition by high levels of viremia,51 or their suppression by CD4+CD25+ regulatory T cells.52
T Cell-Mediated Apoptosis
Both in vivo and in vitro, HIV infection is associated with an activated T-cell phenotype,53-57 increased expression of Fas, enhanced susceptibility to Fas-mediated killing,36,58-63 and increased T cell expressed FasL after T cell receptor stimulation,19,64 suggesting a role for Fas/FasL in HIV- associated AICD.
As CD4+ T cells can upregulate FasL, either by direct in vivo HIV infection or through the effect of virus proteins such as gp120, Tat, or Nef, they can become possible killers of activated Fas-expressing cells, which are found in high numbers in HIV-positive individuals. This hypothesis is supported by the observation that, in vitro, activated CD4+ T cells that express FasL can kill Fas- expressing CD8+ T cells independent of antigen recognition.9,65 Also, HIV-specific CTLs are potential effectors for killing Fas-expressing activated lymphocytes.10 An MHC class-I-restricted CTL clone specific for Nef, derived from an HIV-positive individual, is able to mediate both perforin- and Fas-expressing cell deaths.66 The observation that retinoic acid inhibits the expression of FasL and the resultant CD4+ T cell apoptosis ex vivo67,68 further supports a causal role for Fas-FasL interactions in the CD4+ T cell death that is induced by HIV. Therefore, some of the HIV- specific effectors might be harmful to the immune system of an HIV-positive individual through the FasL-dependent destruction of uninfected Fas-expressing T cells, which are induced by nonspecific, HIV-driven immune stimulation. Altered differentiation of CTLs might be linked to increased apoptosis, as differently differentiated CD8+ T cell subsets have been found to have different susceptibilities to Fas-induced apoptosis.69 Accordingly, rescue from apoptosis of the Bcl- 2low CD8+ T cell subset, which is detected in the lymph nodes and blood of individuals that are chronically infected with HIV,70,71 can be induced by incubation with interleukin (IL)-2 and IL- 15.72,73 IL-15, which is mainly produced by M0, has many activities in common with IL-2: it is required for the survival of memory CD8+ T cells, natural killer (NK) cells, and NK-T cells, and it modulates immune functions of PBMCs from HIV-positive individuals by increasing NK-cell cytotoxicity and enhancing T cell proliferation in response to mitogens and opportunistic antigens.73 In addition, IL-15 is a potent survival factor in the prevention of AICD of CD4+ and CD8+ T cells from HIV-positive individuals and functions by upregulating the expression of Bcl-2.73 Under the same experimental conditions, IL-2, but not IL-10 or -12, has a protective effect on CD4+ and CD8+ T cells.73 However, IL-15 is not able to prevent Fas-induced apoptosis of T cells in vitro,13 in contrast to its in vivo effect in a mouse model.74 IL-12 and -10 partially inhibit Fas-induced apoptosis of CD4+ T cells and CD8+ T cells from HIV-positive individuals, respectively.73,75 IL-15 could, therefore, be an effective promoter of innate immune responses by contributing to the development and survival of TH1 cells. Accordingly, recent attention has turned toward IL-15 as a possible alternative immunotherapy for HIV-positive individuals.
More on the topic AUTOLOGOUS CELL-MEDIATED KILLING VIA INTERCELLULAR CONTACTS:
- The Paradox of Self-Killing
- Endocardial Spindle Cell Proliferation
- CASE 96: Limitations on Killing a Daughter
- CONTENTS
- Killing a Spaniard
- Killing the Family
- CASE 51: Killing the Adulterer...
- KILLING OF UNINFECTED (BYSTANDER) T CELLS BY HIV
- 1,25(OH)2D3-VDR Mediated Transcriptional Regulation of Immune Related Genes
- SOMATIC CELL COUNTS