BIOLOGY OF VPR
Nuclear Entry by Preintegration Complexes
An unsolved puzzle for many years is how HIV causes productive infection in macrophages when nuclear breakdown does not occur as part of ongoing cell division.
In contrast to retrovirus, such as murine moloney leukemia virus, a unique feature of lentiviruses such as SIV and HIV is the ability to permit productive infection in cells that do not exhibit breakdown of the nuclear membrane. One of the first activities of Vpr was the recognition of its involvement in transport of the HIV-1 preintegration complex (PIC) to the nucleus.33-35 To a large extent, this discovery helped clarify the essential role of Vpr for productive infection of macrophages,36,37 because at the time, it was reported that Vpr was dispensable for viral replication in T cells. A number of studies identified features of Vpr that are related to HIV-1 nuclear translocation. One possibility may be that Vpr can enter the nucleus and localize to the nuclear envelope.21 Alternatively, disruption of nuclear envelope integrity by dynamic herniations may allow entry of large PIC complexes.38However, for most proteins targeted for nuclear import, the universal feature is the use of the classical nuclear localization signal (NLS). The NLS is recognized by a family of karyopherin proteins known as importins. Importin-α binds to the NLS, which, together, further interact with importin-β. The importin-α-β complex docks to a nuclear pore complex and is then shuttled across the nuclear pore via a number of energy-dependent steps. Because Vpr does not possess a conventional NLS, it was suggested that it binds directly to importin-α21,35 and, therefore, imitates the action of importin-β.39,40 This form of nuclear import mimickery was described in the transport of proteins that bypass the requirement for importin-α, using importin-β directly.
The precise targeting sequence for this form of transport was not mapped, although sequences rich in arginine residues seem to be a common feature.41 In addition, Vpr’s access to the nucleus is not inhibited by traditional NLS blockers, consistent with the possibility that Vpr may use this alternative form of transport that is independent of the known importin pathway. Controversy surrounding this model has been proposed, because Vpr was shown to be small enough to passively diffuse across the nuclear pore complex.42 Recent work also indicates that Vpr possesses a nuclear export signal (NES)43 to allow exit of Vpr back into the cytoplasm for subsequent PIC incorporation. This property is thought to facilitate infection of macrophages and other nondividing cells. However, one group reported that Vpr was dispensable for ex vivo productive infection of resting T cells.44 These confounding data indicated that the effect of PIC import may differ depending on the target cell type.Induction of Cell-Cycle Arrest
Control of cell-cycle progression is a complex process involved in growth, proliferation, development, DNA damage, and repair. Numerous regulatory proteins ensure that specific events are coordinated to guarantee genomic stability for the proper production of two daughter cells. The major checkpoint regulators in the G2-mitosis interface are the cyclin-dependent kinases, cdc2, and cyclin B1. Activation of cdc2-cyclin B1 permits progression from G2 to mitosis, whereas inactivation of the complex results in G2 cell-cycle arrest. In the inactive cdc2-cyclin B1 complex, cdc2 is phosphorylated on three residues, Thr14, Tyr15, and Thr161. The kinases responsible for Thr14 and Th15 phosphorylation are Wee1 and Myt1, respectively, and the cdk activation complex is responsible for phosphorylation at Thr161. When activation of cdc2-cyclin B1 is required, Thr14 and Tyr15 are dephosphorylated by the cdc25c phosphatase. Similar to yeast, the activities of Wee1 and cdc25c are also governed by their phosphorylation status, such that Wee1 phosphorylation renders it inactive and, conversely, cdc25c active.
Cdc2-cyclin B1 kinase targets both Wee1 and cdc2, which provides a positive feedback amplification that reinforces cdc2 dephosphorylation and irreversible entry into mitosis (reviewed in Amini, Khalili, and Sawaya45).Precisely how Vpr mediates cell-cycle arrest is under debate. Are the effects of Vpr epistatic to cell-cycle regulators, or are their effects secondary to a DNA damaging response? In overexpression experiments, a correlation was observed between the level of Vpr and the hyperphosphorylation state of cdc2 and hypophosphorylation state of cdc25c.30,46 Another possible explanation of how Vpr induces G2 arrest comes from one study in which overexpression of Vpr caused marked alternations and breaks in the nuclear lamin structure. This loss of nuclear envelope integrity would conceivably disrupt the segregation of the cdc2 and cyclin B1 in the nucleus and cytoplasm. Physical stress associated with nuclear disruption may also trigger a DNA damage response that results from incomplete DNA synthesis.47 Mutational analysis demonstrated that an intact C-terminal Vpr domain is essential to mediate G2 arrest, as truncations or point mutations in this domain fail to induce cellcycle arrest.7 A summary of Vpr’s action on the nuclear function during the cell cycle is depicted in Figure 7.2.
Molecular Targets of Vpr
Vpr has multiple activities and interacts with host machinery involved in these processes, such as cell-cycle regulators and proteins that control the apoptotic response. Here, we describe a set of potential interactors of Vpr and discuss the relevance of such interactions.
Early reports indicate that activation of HIV-1 long terminal repeat (LTR) or other heterologous viral promoters requires the activity of Vpr (reviewed in Cohen48), in association with the host transcription factor SP1. Although a direct interaction between Vpr and SP1 was not found, Vpr associates with SP1 responsive elements in coimmunoprecipitation and gel shift assays.49 This association is mediated by the second -helix of Vpr.
Other transcription factors that can associate with Vpr include TFIIB, as shown by GST pull-down assays50 and by acting as an adaptor molecular to bridge the co-activators p300∕cAMP-responsive element-binding (CREB) protein.51 Both in vitro and in vivo binding assays confirm that the interaction of Vpr and p300 depends on the third -helix of Vpr.51In a large-scale yeast two-hybrid screen with wild-type or mutant Vpr molecules as bait, several additional host proteins were found to interact with Vpr. Among these proteins was a kinase, hVIP∕mov34, a homologue of yeast mov34 that belongs to a family of proteosomal and transcriptional regulators.52 The binding of hVIP∕mov34 requires the C-terminal portion of Vpr. However, the relevance of this interaction on cell-cycle arrest is not known. In another yeast two-hybrid screen, two groups, the human homologue of yeast rad23 and HHR23A, bound to Vpr through its C-terminal,53-55 which when overexpressed suppressed the cell-cycle effect of Vpr.55 However, using mutational analysis, other investigators did not observe this effect of HH23RA on binding and cellcycle arrest.56 Several other Vpr-interacting molecules were described. For example, IκB and STAT5β binds Vpr and may have consequences on immune activation and its ensuing humoral and innate responses.56-58
Using a mutant form of Vpr, L64A, as bait, several human 14-3-3 proteins were identified as potential Vpr-binding partners. The 14-3-3 proteins comprise a family of nine isotypes that bind to phosphorylated serine/threonine residues and regulate the activities of their target through subcellular
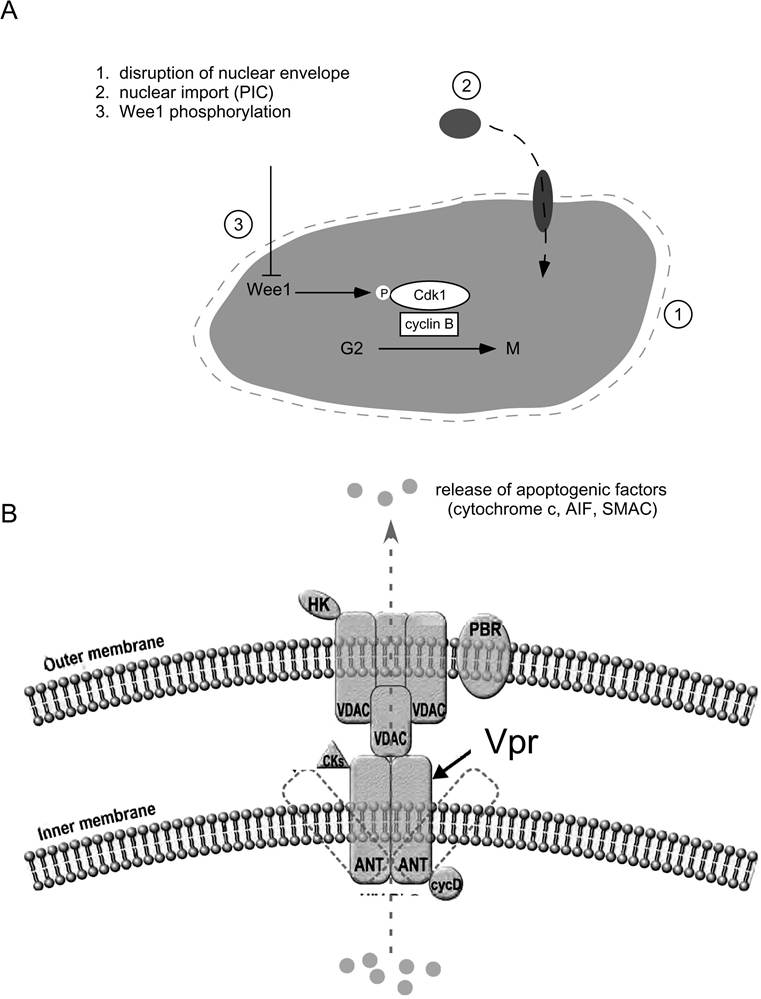
FIGURE 7.2 Major nuclear and mitochondrial targets of Vpr. (A) Vpr disrupts nuclear envelope structure by invoking dynamic herniations, allowing direct import of preintegration complexes and disturbing cellular localization of cell-cycle regulators.
Vpr also targets nuclear pore complexes by mimicking importin proteins and bypassing the requirement for a nuclear localization signal. Wee 1 kinase is regulated by its level of phosphorylation and its subsequent activity on cdc2. Expression of Vpr correlates with hyperphosphorylated Wee1 and G2 arrest. (B) Vpr induces cell death through its interaction with the adenine nucleotide translocator on the inner mitochondrial membrane. This leads to release of apoptogenic factors including cytochrome c, AIF, and SMAC.localization and stability. The role of 14-3-3 proteins in modulating cell-cycle progression was well documented. They regulate the activity of Cdc25C activity59-62 and bind to Wee1 kinases to increase stability. In addition, Chk1/2 kinases are activated by 14-3-3 by sequestering in the nucleus.63 Conversely, 14-3-3 proteins inactivate phosphorylated cdc2 and cyclin B1 by exporting them into the cytoplasm.64 The 14-3-3- attenuated Vpr induced cell-cycle arrest, and lack of the σ-isotype reduced the effect of Vpr. This effect was dependent on binding to the C-terminal portion of Vpr and correlates with the role of Vpr in modulating the activities of cell-cycle regulators. Therefore, the reported effect of Vpr on cell-cycle arrest may, in part, be the result of its association with 14-3-3.
In addition, Vpr was found to interact with a number of multifunctional cellular proteins. The molecular basis and biological consequences of these interactions were not clarified, but they offer new insight into the action of Vpr during HIV infection. The conservation of a proline-rich motif at residues 14 and 35 in the N-terminal of Vpr suggests the possibility of protein-protein interactions. This domain exhibits a high degree of c/T-conformations of the imidic bond. One study suggested that a host peptidyl-prolyl isomerase (PPIase) regulates the interconversion of the imidic bond in the N-terminal of Vpr.65 Cyclophilin A belongs to the PPIase family and is a highly abundant protein found to be associated with capsid proteins of HIV-1 virions.
Previous work supported that cyclo- philin A functions to support the formation of infectious virions.66 This study demonstrated, in pulldown experiments, that Vpr was found to interact with cyclophilin A and, importantly, that cyclo- philin A was required for the de novo expression of Vpr. This interplay was dependent on the N- terminal proline residues. In the absence of cyclophilin A, Vpr incorporation into virions is decreased, and cell-cycle arrest is impaired.Vpr was reported to interact with the DNA repair enzyme, uracil DNA glycosylase (UNG). This interaction was identified in a yeast two-hybrid screen. UNG can be incorporated into viral particles, and this correlates with a reduced rate of reverse transcriptase mutation in the viral genome.67,68 This may select for increased fitness of progeny. Another way in which Vpr is thought to disrupt the DNA/RNA processing machinery is through the lys-tRNA synthetase product, tRNAlys. tRNAlys functions as the primer for the reverse transcriptase enzyme. The N-terminal of Vpr is thought to interact with lys-tRNA synthetase and thereby disrupt initiation of reverse transcription of the viral genome.69
An important step forward toward understanding the apoptosis-inducing effects of Vpr was the observation that Vpr can directly interact with the mitochondria in yeast. However, because yeast lack core components of the apoptotic machinery, it was unclear what the role of mitochondria played during Vpr-induced eukaryotic cell death. In mammalian cells, an apoptotic response initiated through the intrinsic pathway results in mitochondria that lose membrane potential. Although there is still considerable debate as to whether the mitochondria permeability transition pore complex (PTPC) is in an open or closed conformation, the resulting release of cytochrome c induces a cascade of events leading to the eventual cleavage of effector caspases and subsequent cell death. The mitochondria PTPC is a multiprotein complex that includes the adenine nucleotide translocator (ANT) found on the inner mitochondria membrane. Synthetic Vpr peptides directly bind to ANT, and this correlates with a decrease in mitochondria membrane potential and release of cytochrome c, whereas Vpr mutant peptides do not have this effect.11,70 Subsequent experiments demonstrated that the Vpr-induced apoptosis can be blocked by HIV-1 protease inhibitors, cyclophilin A inhibitors, and bonkregic acid, laying further support for ANT as a molecular target for Vpr.11,70,71