HIV KILLING OF INFECTED T CELLS
How do HIV-infected cells die? A number of general pathways promoting cell death have been dissected in detail. We will discuss how HIV infection, as well as the actions of isolated viral factors, influences these cell death pathways.
Direct Killing by Apoptosis and Other Mechanisms
Precise characterization of HIV-mediated cell death has proved rather difficult. Although many studies have presented evidence for direct HIV-mediated death in T cell lines or primary T cells, it is often hard to ascertain whether the dead cells correspond to infected cells, bystander cells, or a combination of both. Not surprisingly, the literature is replete with conflicting reports. The few studies that have characterized HIV-infected cells at the single-cell level with reporter genes and flow cytometry have implicated apoptosis as the major mechanism of cell death, including hallmarks of this death pathway such as loss of mitochondrial membrane potential,11 phosphatidylserine accumulation on the exterior plasma membrane,12 and fragmentation of DNA.13,14 These observations are supported by some studies in HIV-infected cultures15-17 but not by others.11,18-21 The reasons for these experimental disparities remain unclear. These studies do agree that death receptor-mediated signaling through death adaptors involving caspase-8 activation does not seem to play a major role and that broadly acting caspase inhibitors often fail to protect infected cells from death.11,14,19
In summary, HIV infection in many cellular systems leads to phenotypic changes consistent with apoptosis, yet classic caspase cascades may not play a prominent role. HIV-induced cytotoxicity probably depends on the expression levels of various viral and host proteins that establish a cellular threshold for apoptosis. In cells with a high threshold, HIV infection may be so damaging that death results, regardless of whether the classical apoptotic signaling pathways are detectably induced.
Cytopathicity of the HIV Envelope Protein Involves Both CD4
and the Chemokine Co-Receptors
The HIV envelope (Env) protein is a type I transmembrane protein, initially translated in the endoplasmic reticulum (ER) as a 160 kDa molecule (gp160). In the ER, gp160 is cleaved by the cellular protease furin. The resulting transmembrane gp41 protein contains the fusion peptide, whereas the noncovalently associated surface gp120 protein contains domains that sequentially interact with CD4 and one of two principal HIV co-receptors, CCR5 or CXCR4. These gp120 interactions trigger a conformational change in gp41 structure that exposes the fusion peptide, promoting its insertion into the target cell membrane. Importantly, gp120 and gp160 associate with CD4 at the plasma membrane and within the endoplasmic reticulum, respectively. In fact, interactions in the ER are important for the viral life cycle: they retard surface expression of CD4 maturation and act in concert with a second HIV protein, Vpu, which promotes polyubiquitylation of CD4 and its degradation by the 26S proteasome. Because the interaction between gp120 and gp41 is noncovalent, gp120 can be shed from the surface of either HIV-infected cells or virions.22,23
Env was the first viral protein shown to promote cell death.24,25 Three models emerged to explain its cytopathic effects. The first proposes that gp120 transiently interacts with CD4 and the HIV coreceptor on target cells and triggers a set of intracellular signaling events that promotes cell death. The source of gp120 can be the virion surface, infected cells, or shed gp120. Because CD4 is effectively downregulated from the surface of infected cells by the combined action of the HIV proteins Env,26,27 Nef,28,29 and Vpu,30,31 this mechanism would presumably target bystander cells expressing CD4 (Figure 13.3 and Figure 13.4).
A second model, also principally related to bystander killing, holds that expression of gp120/41 on the surface of infected cells can mediate fusion between infected and uninfected cells, leading to syncytia formation (Figure 13.3).24,25 Of note is that syncytia appear to occur principally in T cell lines in vitro,32 although some evidence for syncytia has been documented in vivo?3-31
In the third model, Env mediates “single-cell killing,”38-40 a phrase used to describe cell death occurring in HIV-infected cultures in the absence of syncytia formation.
In fact, single-cell killing, syncytia formation, and the promotion of viral replication are genetically separable events within Env.32,38 In HIV-infected primary cell cultures and some T cell lines, single-cell killing is the most prominent form of cell death.32Three models have also been proposed to explain how Env mediates single-cell killing (Figure 13.2A). They all agree that Env must interact with CD4 but disagree over the consequences of this interaction or where in the cell this interplay occurs. Nomoto and colleagues hypothesized that unprocessed gp160 interacts with CD4 in the ER, leading to apoptosis. Although the regions of gp160 and CD4 necessary for this action, as well as the mechanism, are unclear, complexes of unprocessed gp160 and CD4 were correlated with a disruption of nuclear pores and disturbances in calcium signaling.41-45 Sodroski and colleagues favor a second model that suggests that intracellular fusion events may be facilitated by interactions between gp120∕41, CD4, and the appropriate co-receptor in the secretory pathway.39,46-49 These intracellular fusion events damage cellular membranes and lead to cell death reminiscent of necrosis. This phenotype was observed in HIV-infected cultures and in cells expressing the HIV envelope together with CD4 and the appropriate co-receptor. Two key findings support this model: single-cell killing is resistant to soluble CD4, and the viral envelope must not only be able to interact with CD4 but must also possess a functional fusion peptide.39,46-49 A third model supported by Corbeil and colleagues states that surface-expressed complexes of gp120∕41 and CD4 are toxic and that the cytoplasmic tail of CD4, but not its ability to signal through the tyrosine kinase Lck, is important for this response.50-52 In contrast to the findings of the Sodroski group, these authors report that Env- mediated killing can be inhibited by soluble CD4.46,50-52 Thus, in summary, although Env is one of the most potent mediators of HIV-induced cell death, the molecular mechanism underlying its action remains highly controversial.
Vpr Mediates Apoptosis by Directly Targeting the Mitochondria
Expression of viral protein R (Vpr) in a variety of systems induces apoptosis. Perhaps most telling is that genetic studies53-55 and studies of Vpr alleles from long-term nonprogressors56,57 have demonstrated that Vpr is a key determinant of cytopathicity. Expressed alone, Vpr is cytotoxic in systems as disparate as yeast,58 murine T cells,59 and human T cells,60-63 although some studies
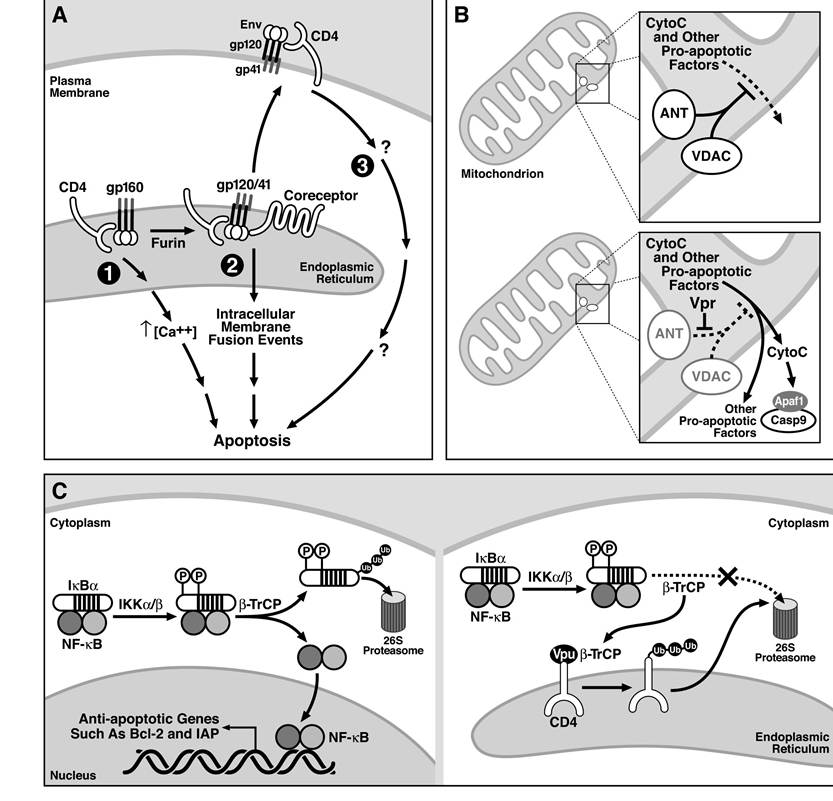
FIGURE 13.2 Models underlying direct killing by HIV Env, Vpr, and Vpu. (A) The three models of Env- mediated cytotoxicity, including (1) gp160 binding to CD4 in the ER leading to cytotoxic signaling, (2) intracellular fusion events involving Env∕CD4-co-receptor interactions, and (3) gp120 signaling from the cell surface through HIV receptors, are outlined and discussed in the text. (B) Vpr directly interacts with and inhibits the activity of ANT, a key component of the machinery needed to maintain the integrity of the mitochondria. (C) In the absence of Vpu, β-TrCP plays a key role in degrading IκBα, which allows for NF-κB mediated activation of antiapoptotic genes. In the presence of Vpu, β-TrCP binds to and is sequestered by Vpu, thereby inhibiting the normal degradation of IκBα and activation of NF-κB. NF-κB regulates the expression of several key antiapoptotic genes.
have suggested that this phenotype depends on either the levels of Vpr64’65 or the activation state of the cell.66 This is an important point, because Vpr is detectable in patient serum,67 in part, because it can cross biological membranes in an energy-independent manner.68 Thus, at sufficient concentrations, Vpr could mediate the killing of infected and bystander cells (Figure 13.4). Vpr-induced cell death seems to be apoptotic: it typically correlates with activation of caspases, loss of mitochondrial membrane potential, release of cytochrome c into the cytoplasm, phosphatidylserine exteriorization, and hypodiploidy.57’60’62’63’69-72 Activation of caspases seems to play a significant
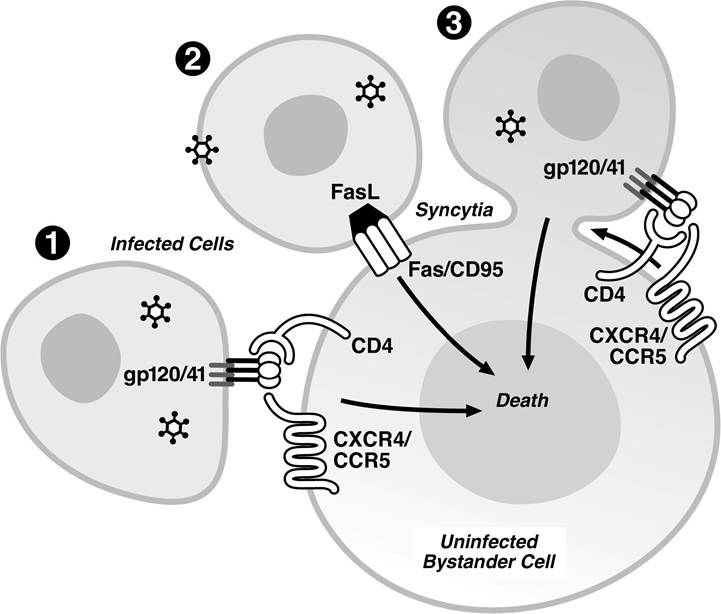
FIGURE 13.3 Possible mechanisms of bystander CD4+ T cell killing by direct cell-cell interaction.
(1) Interaction of Env expressed on the surface of HIV-infected cells with CD4 and a co-receptor expressed on the surface of the uninfected T cells. (2) Engagement of Fas receptors on uninfected cells by FasL expressed on the surface of the infected cells. (3) Syncytia formation induced by the interaction of HIV Env expressed on the surface of infected cells and the CD4/co-receptors present on bystander cells promoting cell-to-cell fusion.role in Vpr-mediated toxicity, because Vpr-mediated killing can be inhibited by broadly acting caspase inhibitors.57,61,62’72
Although the connection between most other cytotoxic HIV genes and the cellular death machinery may be indirect, Vpr directly targets one of two key proteins that regulate mitochondrial membrane potential (MMP). In a cell-free system, Vpr effectively disrupts MMP, facilitating the release of cytochrome c.η2 MMP is regulated by the permeability transition pore complex that comprises the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane leaflet and the adenine nucleotide translocator (ANT) located on the inner leaflet. These proteins interact at sites where the two membranes are juxtaposed. Vpr physically interacts with ANT with nanomolar affinity, and the interaction leads to changes in membrane permeability.72’73 Consistent with the finding that Bcl-2 antagonizes the proapoptotic affects of Vpr, Bcl-2 decreases the affinity of the interaction of Vpr with ANT. These studies suggest that Vpr directly targets and destabilizes a key component of the mitochondria responsible for regulating membrane potential (Figure 13.2B).73 Surprisingly, a recent study by the same group indicates that Vpr-induced toxicity occurs independently of the caspases and other catabolic hydrolases that are normally activated after cytochrome c release from the mitochondria.74 As noted earlier for HIV-mediated direct killing, this is probably another example in which, even though the dominant death pathway is inhibited, sufficient toxic effects are generated, perhaps as a result of bioenergetic catastrophe, and cell death ensues.
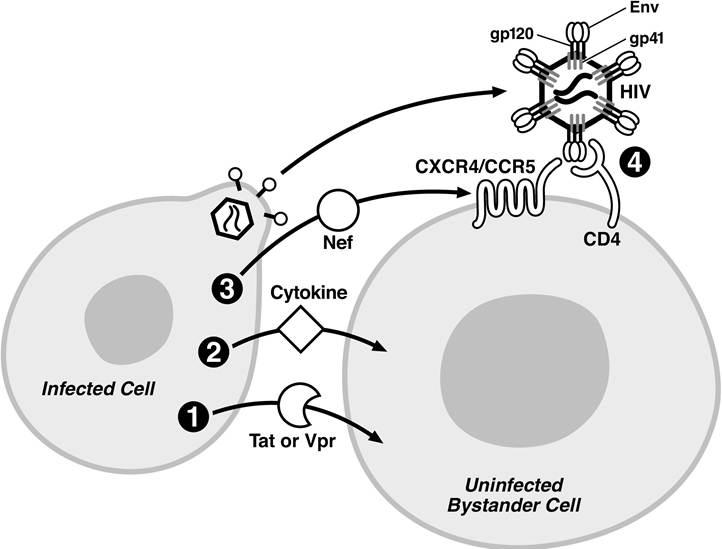
FIGURE 13.4 Possible mechanisms of bystander CD4+ T cell killing by soluble factors. (1) Cytopathic effect of soluble viral factors (Tat or Vpr) entering bystander cells due to their protein transduction properties. (2) Production of cytotoxic cytokines by infected cells, altering viability of bystander cells. (3) Binding of extracellular Nef to CXCR4, triggering cytotoxic effects in the bystander cells. (4) Interaction of gp120 presented on HIV virions budding from infected cells with CD4 and co-receptors present on the surface of the bystander CD4+ T cells. The HIV virion is not depicted to scale.
Vpu Contributes to Apoptosis by Inhibiting the Antiapoptotic
Effects of NF-κB
Although far fewer studies have reported proapoptotic effects of Vpu than those of Env and Vpr, a recent genetic screen has yielded intriguing results. Strebel and colleagues identified Vpu as a proapoptotic gene after screening HIV mutants lacking individual genes for their ability to induce apoptosis in Jurkat T cells. They further confirmed the role of Vpu as a proapoptotic gene during infection of primary CD4+ T cells.53 Although it is clear in these experiments that Vpu functions as a proapoptotic gene, infections with HIV viruses lacking Vpu still induce cytopathic effects, supporting a role for other HIV genes as well.54,75
The ability of Vpu to promote apoptosis likely involves inhibition of NF-κB signaling (Figure 13.2C). Activation of NF-κB requires phosphorylation, ubiquitylation, and degradation of the NF-κB inhibitor IκBα.76 IκBα is ubiquitylated by the same E3 ubiquitin ligase, β-TrCP,77-82 employed by Vpu to ubiquitylate CD4.83 By competing for β-TrCP, Vpu inhibits IκBα degradation, which, in turn, prevents NF-κB from promoting the expression of various antiapoptotic genes. The result is that Vpu is proap- optotic.84,85 What is unclear from this study is why the proapoptotic potential of Vpu emerged only recently and not in similar previous studies.54,75 Perhaps the endogenous levels of β-TrCP, IκBα, NF-κB, or NF-κB-induced antiapoptotic genes in some cell types confer greater or lesser sensitivity to this pathway.
HIV Protease as a Mediator of Direct Cell Killing
HIV protease (PR) is toxic when expressed in Escherichia coli, Saccharomyces cerevisiae, or mammalian fibroblast cell lines.86-88 Although PR cleaves a variety of host substrates in vitro,89-96 few have been demonstrated to be responsible for the toxicity of PR. Two possible exceptions are Bcl-297 and caspase-8.98 Both are cleaved in infected T cell lines, although neither study has convincingly demonstrated similar effects in primary CD4+ T cells. Perhaps this result is not altogether surprising, as enzymatically active PR in an HIV-infected cell would most likely be restricted to the plasma membrane, whereas Bcl-2 is principally localized at the mitochondria. Of note is that a recently described PR mutant supports normal viral replication but lacks cytotoxic effects.99 It will be interesting to investigate the ability of this mutant to inactivate Bcl-2 and to activate caspase-8 as well as other proapoptotic factors in the cell. Although PR displays cytotoxic properties, the biological significance of these findings in HIV-infected cells remains unclear.
Tat Increases the Sensitivity of Infected Cells to Apoptosis
Tat has attracted considerable interest as a proapoptotic factor for two reasons. First, like Vpr, Tat contains a basic protein transduction domain that can promote its transit across biological membranes. Thus, Tat can promote the death of both infected and uninfected cells (Figure 13.4).100-103 Second, Tat has been reported to interact directly with a number of proteins involved in transcription, including the RNA Pol II elongation factor, pTEF-b104-106, the acetyltransferases p300107-109, pCAF108, CREB-binding protein107, and the transcription factor Egr.110 Consequently, Tat may exert a wide range of effects on host gene expression that will make it difficult to identify a single proapoptotic mechanism. Whereas other HIV proteins directly interact with the apoptotic machinery of the host, Tat may modulate apoptosis by altering the balance of pro- and antiapoptotic gene expression.
A number of different approaches, including treatment of cells with soluble Tat or stable expression of Tat within cells, have shown that Tat either is a proapoptotic factor or renders cells more sensitive to other proapoptotic factors. However, no clear consensus on the underlying mechanism has emerged. One model suggests that Tat-mediated activation of cyclin-dependent kinases underlies its proapoptotic activity. Inhibition of these kinases reduced Tat-mediated apoptosis.111 Another model suggests that the proapoptotic activity of Tat is associated with its ability to alter the sensitivity of cells to external death signals. This notion is controversial. Specifically, although various groups have documented increased levels of CD95L expression112,113 or increased sensitivity to Fas-mediated death,75,114 others have failed to detect these changes.115,116 In one report, cell death in HIV-infected T cell cultures was prevented by neutralizing CD95 or Tat.113 The ability of Tat to increase CD95L expression levels was associated with the activation of the transcription factors NF-κB117 or Egr.110 Another potentially key change elicited by Tat is increased expression of caspase-8.75 The relevance of these findings to HIV- infected T cells remains unclear, because increases in CD95L expression or Fas sensitivity were not detected in such cultures.14,118
In light of the controversy surrounding these findings, the proposed actions of Tat further downstream merit consideration. Tat can indirectly alter the redox state of the cell by downregulating Mn superoxide dismutase.119 This leads to increased levels of reactive oxygen intermediates115 that may exert proapoptotic effects. In addition, Tat has been proposed to alter the regulation of Bcl-2 family members, although both increases120-122 and decreases116,123 in the expression of the antiapo- ptotic Bcl-2 protein have been described. Levels of the proapoptotic proteins Bax116 and Bim124 are increased. These findings are consistent with increased levels of activated caspase-9124 and caspase-3123 in Tat-expressing cells. Finally, the use of pan-caspase inhibitors or a caspase-3 inhibitor prevents Tat-induced cell death, implicating activation of a classical cysteine protease cascade in Tat-induced cell death.124
Of note, Tat has also been reported to exert antiapoptotic effects. This claim is based on functional studies120,122,125-127 and increases in the expression of Bcl-2.120,122,128 The root of the discrepancy in these contrasting results remains unclear but likely reflects differences in the cell types analyzed and perhaps in the levels of Tat expression achieved.
More on the topic HIV KILLING OF INFECTED T CELLS:
- CLINICAL SIGNIFICANCE OF THE CELL DEATH INDUCED BY PR
- Killing a Spaniard
- APOPTOSIS IN ANIMAL MODELS OF HIV-1 DISEASE
- INTERFERENCE WITH THYMIC FUNCTION
- CD8+ T LYMPHOCYTES IN HIV-1 INFECTION
- HIV INFECTION OF THE CD8+ T CELLS: CONTROVERSY AND CLARIFICATION
- NEUROLOGICAL DYSFUNCTION ASSOCIATED WITH HIV-1 INFECTION
- General management of HIV-infected people
- 24 HIV in Pregnancy
- HIV INFECTION OF THYMOCYTE SUBSETS