INTRODUCTION
The progressive loss of CD4+ T cells is a defining clinical feature of AIDS resulting from infection with the human immunodeficiency virus (HIV). CD4+ T cell depletion in HIV-infected individuals almost certainly is a multifactorial process.
Remarkably, the mechanisms underlying this phenomenon remain poorly understood. In this chapter, we will review the mechanisms implicated in both the direct death of HIV-infected cells and the indirect HIV-mediated death of uninfected CD4+ T cells, often termed “bystander killing.”On the basis of morphological changes, two types of cell death have been described: apoptosis and necrosis. In necrotic cells, swelling of the cytoplasm leads to destruction of organelles and eventual rupture of the plasma membrane. The mechanism of cellular necrosis is not well understood and is often evoked as a mechanism only after a role for apoptosis has been excluded. Morphologically, apoptotic cells are characterized by cell shrinkage, intact organelles, and the condensation and fragmentation of DNA. Many of the molecular mechanisms underlying apoptosis have been identified and shown to be regulated at multiple levels.1-3 Please note that although the terms apoptosis and necrosis are useful descriptions, several recent papers have emphasized that these pathways may overlap, making their distinction more difficult.4-5
The apoptotic signaling cascade can be initiated by extracellular or intracellular signals (Figure 13.1). Extracellular signals frequently involve the binding of members of the tumor necrosis
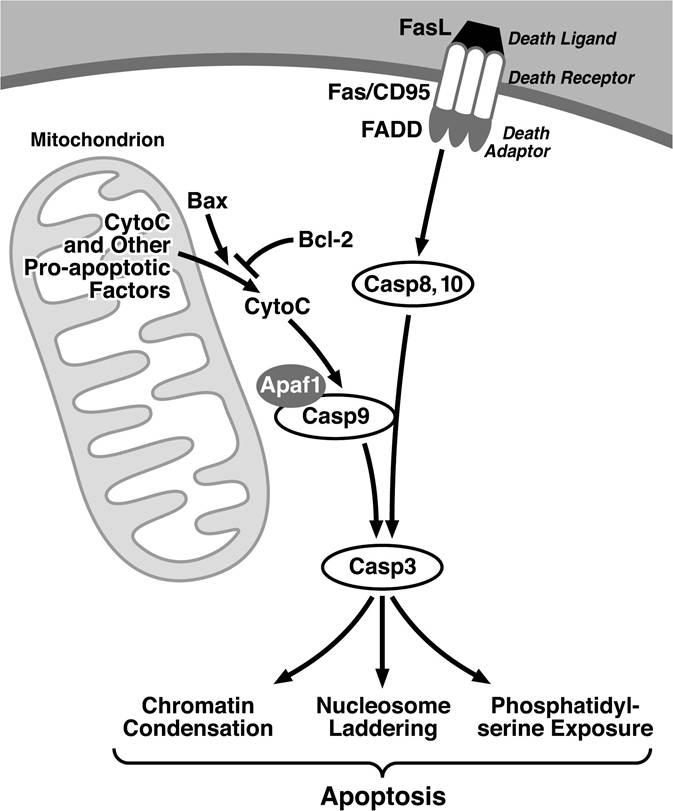
FIGURE 13.1 Extrinsic and intrinsic signaling pathways underlying apoptosis. Extrensic signals from death ligands are transmitted by trimerized death receptors such as Fas promoting recruitment of death adaptors culminating in the activation of the initiator caspases, caspase-8, and -10.
This leads to the cleavage of the executor caspase, caspase-3, which either directly or indirectly underlies many of the phenotypes observed during apoptosis. Caspase-3 also can be activated by a complex of caspase-9 and Apaf1, which is activated when cytochrome-C is released from the mitochondria. Release of cytochrome-C and other apoptosis-inducing molecules from the mitochondria is both negatively and positively regulated by various Bcl-2 family members including Bcl-2 and Bax, respectively.factor (TNF) superfamily to a set of death domain-containing receptors.6 The death domain motifs in the cytoplasmic tail of these receptors inducibly bind to a set of adaptors that, in turn, mediate the activation of a family of cysteine proteases, known as caspases.7 In particular, caspase-8 and caspase-10 interact with and are activated by death receptor adaptors that initiate a cascade of proteases, culminating in the activation caspase-3. By direct or indirect mechanisms, active caspase-3 causes many of the features of apoptosis. Thus, activation of caspase-3 is viewed as “a point of no return” in the apoptotic pathway.
Intracellular proapoptotic signals, such as DNA damage, are primarily coordinated by factors residing in the mitochondria and lead to the activation of caspase-3. Key among these mitochondrial factors is cytochrome c. Release of cytochrome c from the mitochondria promotes activation of caspase-9 involving the action of Apaf-1. Caspase-9 activation leads, in turn, to the activation of caspase-3. Release of cytochrome c and other mitochondrial proteins is controlled by Bcl-2 family members8,9 that can function as either pro- or antiapoptotic factors. Proapoptotic Bcl-2 family members, such as Bax and Bak, are inhibited by heterodimerization with antiapoptotic Bcl-2 family members, such as Bcl-2 and Bcl-xL. The relative expression levels of these proteins within a cell establish a threshold for apoptosis. NF-κB is one of the key transcription factors that controls the levels of antiapoptotic molecules.10