NEF SIGNALS THAT STIMULATE ANTI- AND PROAPOPTOTIC PATHWAYS
Survival Strategies of Viruses: Antiapoptosis and Immune Evasion
In recent years, we have started to understand how important survival strategies are for invading pathogenic viruses, in particular, when they intend to establish a chronic infection.22,45,46 In fact, in a chronic phase of a viral infection, when viral replication is often barely detectable, these mechanisms may be even more important than viral replication.
Recent work has shown that most viruses have a bivalent defense strategy. The first arm of this strategy relies on numerous mechanisms of immune evasion, which include change of protein conformation and sequence, interference with MHC antigen presentation, modulation or mimicry of cytokine activity, and induction of apoptosis by Fas ligand.22 HIV is equipped with several of these mechanisms, two of which are exerted by the Nef protein (see below). Viral-induced immune evasion alone will not ensure survival of an infected cell until the next virus generation is ready to leave. In expecting a viral infection, the cell has learned to self-destruct by apoptosis as soon as something unexpected occurs (i.e., unscheduled DNA synthesis).Cellular death is induced, in principle, by two different routes. Internal sensors (for example, p53) activate the so-called intrinsic pathway, which initiates a process leading to the ultimate loss of mitochondrial integrity and apoptosis. Proteins of the Bcl-2 family (Figure 8.2) regulate this process. Proapoptotic members of this family (Bad, Bax, Bak, Bid, and others) form heterodimers with and thereby inactivate pro-survival members of the same family (Bcl-2, Bcl-Xl, Bcl-w, and others). In antiapoptotic signaling, the proapoptotic effectors such as Bad are phosphorylated on specific serine residues that release Bcl-2 for pro-survival activity by blocking Apaf-1-mediated activation of initiator caspase-9.
The signaling pathway that leads to Bad phosphorylation usually starts with the activation of PI3 kinase, through ligation of cytokine or growth factor receptors. Downstream, PI3 kinase activates the Akt serine/threonine kinase that directly phosphorylated the Bad protein. As discussed above, this mechanism may not apply to T cells (see also below). The middle T antigen of polyomavirus was found to activate this pathway for antiapoptotic signaling.47 Other viruses, particularly lymphotropic herpesviruses such as Epstein-Barr virus (EBV), overexpress a Bcl-2/Bcl-XL homologue to bind and neutralize death-inducing members of the same family. Bcl-2 and Bcl-2 XL prevent the release of mitochondrial cytochrome c and subsequent activation of caspases, presumably by interacting with Apaf-1.48The second, an extrinsic pathway, is initiated through death receptors of the tumor necrosis family (TNF) of receptors transmitting external signals usually provided by immune effector cells (Figure 8.2). So far, five death receptors have been identified: Fas, TNF receptor 1, TRAMP, TRAIL- R1, and TRAIL-R2. Activation of these receptors leads to the recruitment of a death-inducing signaling complex. This complex contains adapter proteins, such as the initiator caspase-8/FLICE, with protein-binding motifs called death domains (DD) and death effector domains (DED). Ultimately, the death-signaling complex leads to the activation of effector caspases and, subsequently, cell death. Viruses, and again the lymphotropic herpesviruses such as Kaposi’s sarcoma-associated herpesvirus (KSHV), overexpress proteins, the so-called viral FLIPs (FLICE inhibitory proteins), which block the recruitment of FLICE and thus the formation of a functional death-inducing signaling complex.48
Interference of Nef with the Death Receptor/Extrinsic Pathway
Signals from the surface through death receptors TNFR1/2 and Fas could potentially stimulate death programs. In HIV infection, this is a realistic scenario.25 First, HIV-mediated upregulation of Fas- ligand (see below) could kill the infected cell in an autocrine fashion through Fas ligation (Figure 8.3).
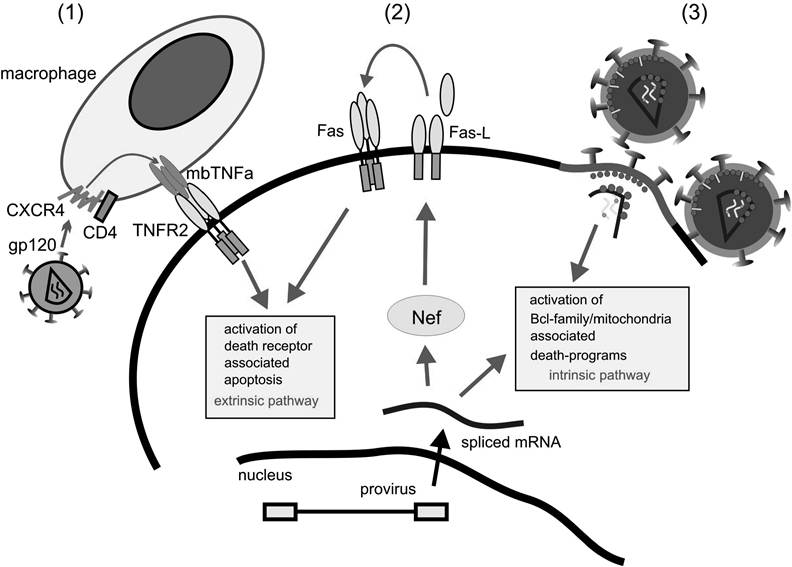
FIGURE 8.3 Apoptosis-inducing signals that threaten the infected cell. Several signals can potentially induce apoptotic cell death in the infected cell. (1) Cross-linking of CXCR4 by HIV gp120 envelope induces the upregulation of membrane-bound TNF on macrophages. This could potentially stimulate the TNF receptor on the infected cells. (2) Nef expression leads to the upregulation of Fas ligand, which could potentially stimulate the Fas receptor in an autocrine fashion. Such a mechanism would lead to the self-destruction of the infected cell. (3) The invading virus during cellular infection likely activates internal sensors that would lead to the activation of the intrinsic death program regulated through the Bcl-2 family.
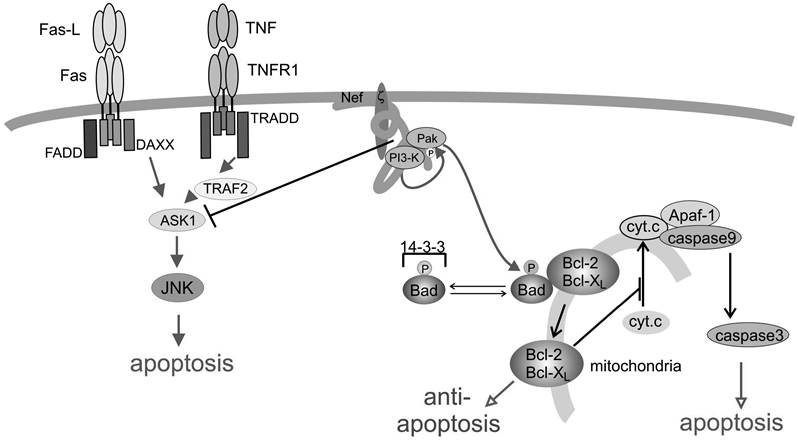
FIGURE 8.4 Nef blocks the extrinsic and intrinsic apoptotic pathways. Nef blocks death receptor-mediated signals through inhibition of ASK1. The serine/threonine kinase ASK1 is an upstream effector of the JNK/p38 pathway and a positive mediator of apoptotic signals. Nef associates with and activates the PI3 and Pak kinases. Subsequently, Pak phosphorylates and inactivates the Bad protein. This liberates Bcl-2 and Bcl-XL to block the release of cytochrome c.
Second, HIV gp120 ligation of CXCR4 on macrophages induces the upregulation of membranebound TNF. This has been shown to induce cell death via the TNFR in CD8 cells (Figure 8.3), which may lead to T cell depletion.49 Because both apoptotic signals, “outside in” by death receptors and “inside in” by unbalanced cellular homeostasis, are regulated by different signaling pathways, the invading virus has to block both routes early and efficiently.
HIV seems to manage this dilemma through functions of the Nef protein. Geleziunas and coworkers50 found that Nef associates with and blocks the activity of apoptosis signal-regulating kinase 1 (ASK1) (Figure 8.4).
ASK1, a MAPKKK, links both the Fas- and the TNFR-mediated signals (by Fas-ligand and TNF) to the downstream JNK/p38 pathways (Figure 8.2). In both instances, ASK1 interacts with the death-signaling complex through association with TRAF2 (TNFR-1/2) and Daxx (Fas).51,52 Although overexpression of ASK induces apoptosis, a transdominant negative ASK1 mutant will block receptor-induced death signals. ASK1 kinase activity is inhibited by thioredoxin (Trx), a redox regulator protein. Only a reduced form of Trx associates with ASK1 and keeps the kinase inactive. A precise mechanism of how Nef activates ASK1 is lacking. However, it seems that Nef blocks the stimulus-dependent release of Trx from ASK1. At this point, it is not clear how this signaling function of Nef connects to the Nef signaling or T cell receptor complex. On the other hand, it has been shown that Nef recruits the Vav protein (see above), which causes the activation and nuclear translocation of the JNK kinase. Therefore, it is conceivable that Nef and death receptor-mediated activation of JNK converge at a level that remains to be determined.Interference of Nef with the Mitochondria-Associated/Intrinsic Pathway
Another report has demonstrated that Nef signaling also interferes with regulation of the intrinsic, mitochondria-associated cellular death program regulated by the Bcl family of proteins. In order to block proapoptotic members of this family, Wolf et al.53 demonstrated that HIV/Nef uses the PI3 kinase pathway, however, in a surprising new variation. Like mTAg, Nef was found to associate with and activate PI3 kinase but not to stimulate Akt, as one would expect, but, rather, to activate the Nef-associated serine kinase Pak. The association and activation of Pak is one of the hallmark signaling functions of Nef that was described early.28 Up to that time, a functional consequence for this interaction was lacking or at least not clearly evident. The Nef-PI3 kinase-PAK complex was able to phosphorylate Bad on serine residues (Figure 8.4), resulting in a block of apoptosis induced by serum starvation and, more importantly, by HIV replication.
As would have been predicted, Nef antiapoptotic signaling increased viral particle release between five- and tenfold from infected Jurkat cells in vitro, which indicated that this mechanism was of particular importance for viral replication. Beyond that, the phosphorylation of Bad through PI3 kinase and Pak suggested an intriguing new signaling pathway emanating from the TCR independent of the Akt kinase.By interfering with the Bcl family, Nef follows the common path of other viruses (e.g., herpesviruses). This shows that this is a particularly efficient way to secure viral interests. Herpesviruses, however, which have large DNA genomes, “bring” and overexpress their own Bcl homologues. Conversely, HIV and polyomavirus, due to their small-sized genomes, had to evolve a different strategy. They manage to induce phosphorylation of Bad, which seems to be comparably efficient. Because polyomavirus and HIV infect different target cells, the signaling pathways leading to Bad inactivation are slightly different. For the same reason, Nef may interfere with ASK1 kinase activity rather than overexpress its “own” protein to block early death reporter signaling (e.g., through vFLIPs).