SKELETAL DYSPLASIA
Skeletal dysplasia is a heterogeneous group of many genetic disorders, all characterized by intrinsic defects in formation of cartilage or bone. Clinical spectrum of these disorders varies from in utero lethal defects to very mild disorders, which may remain undetected throughout the life.
Classification: While many classification systems have been proposed for skeletal dysplasias on the basis of genetic, clinical or pathological characteristics, two broad types are (Table 23.7):
a. Chondrodysplasias, with defective growth of tubular bones and/or spine, e.g. achondroplasia.
b. Osteodyplasia, with defective structural modeling or density of bones, due to:
- Defective collagen, e.g. osteogenesis imperfecta.
- Defective matrix, e.g. mucopolysaccharidoses.
- Defective bone resorption, e.g. osteopetrosis.
Chondrodysplasias are characterized by genetic defects in enchondral bone formation and growth.
TABLE 23.7: Common skeletal dysplasias
A. Chrondodysplasia
Short-limb skeletal dysplasias
- Predominantly rhizomelic
Achondroplasia Hypochondroplasia Thanatophoric dysplasia* Metatrophic dysplasia Chondrodysplasia punctata
- Predominantly mesomelic Camptomelic dysplasia* Mesomelic dysplasia Short-limb polydactyly syndromes*
- Predominantly acromelic Asphyxiating thoracic dystrophy* Ellis-van Creveld syndrome Cartilage-hair hypoplasia Pyknodysostosis
Diastrophic dysplasia Multiple epiphyseal dysplasia
Short-trunk skeletal dysplasias
- With mild micromelia
Spondyloepiphyseal dysplasia Mucopolysaccharoidosis
- With moderate/severe micromelia
Achondrogenesis** Kniest syndrome Spondylometaphyseal dysplasia
Osteodysplasia
Osteogenesis imperfecta**
Osteopetrosis
*lethal at birth or in neonatal period
**some types are lethal at birth
Disproportionate short stature is the hallmark of these disorders, which may be divided into: (a) short-limb dwarfism with disproportionate shortening of limbs as compared to trunk, e.g.
achondroplasia, and (b) shorttrunk dwarfism, e.g. spondyloepiphyseal dysplasias.Short-limb chondrodysplasias are further subclassified as those with: (a) Rhizomelia, i.e. predominant shortening of proximal limb segments like upper arms and thighs, (b) Mesomelia, i.e. predominant shortening of middle segments like forearms and legs and (c) Acromelia, i.e. predominant shortening of distal segments like hands and feet.
Osteodysplasias are characterized by genetic defects in modeling of bones, leading to altered bone density, including two broad subtypes;
a. Disorders with decreased bone density, e.g. osteogenesis imperfecta; and
b. Disorders with increased bone density, e.g. osteopetrosis or pyknodysostosis.
Diagnostic evaluation of skeletal dysplasia is largely based on clinical and radiological features. Important steps in approach to a suspected case are as follows: a. Demonstration of short stature on anthropometric evaluation, though many mild cases may be normal at birth and short stature becomes apparent only in late childhood.
b. Assessment of body proportions: Disproportionate shortening is best assessed by measuring uppersegment : lower-segment ratio (US:LS ratio). Normal US:LS ratio varies with age, being-1.7:1 at birth, 1.3:1 at 3 years and 1:1 at 7 years. Although criteria for individual limb segments are available, Rhizo-/ Meso-/Acro-melia is usually identified by clinical appearance.
c. Clinical features, e.g. age of presentation, family history and presence of coexisting non-skeletal defects provide important clues for detection and classification of diagnosis.
d. Radiological evaluation is often diagnostic and requires complete skeletogram including limb, spine, thorax and skull X-rays in different views.
e. Molecular diagnosis with genetic studies are preferable whenever possible, to decide about the future risks of recurrence.
f. Other laboratory investigations are of a little use, except in osteogenesis imperfecta or MPS.
Some important skeletal dysplasia are as follows:
Achondroplasia: Achondroplasia is the commonest skeletal dysplasia (~1:15,000), clinically characterized by typical short-limb dwarfism and craniofacial abnormalities, apart from other features.
Etiology: Although transmitted as an autosomal dominant disorder, most cases arise from a new mutation in normal parents at FGFR3 gene codon 380, which governs the rate of enchondral bone formation.
Clinically, skeletal manifestations are obvious at birth, while secondary neurological features appear at a later age. Important features include:
• Rhizomelic, short-limb, short stature, i.e. normal sized trunk but relatively shorter extremities, specially in proximal parts, e.g. upper arms and thighs. Hands are broad and short, with a trident-like arrangement of fingers. Bowing of legs is common (Fig. 23.7A)
• Craniofacial abnormalities, e.g. large head, prominent forehead, depressed nasal bridge, hypoplastic maxilla with relative prognathism. Dental malocclusion is common.
Hypotonia is common in earlier years leading to: (i) delayed early motor development, (ii) prominent lumbar gibbus in infancy, gradually replaced by lumbar lordosis as child starts to walk, and (iii) hyperextensible joints, except elbow, where movements may be restricted.
Neurological complications may develop at any age due to skeletal malformations and include: (i) hydrocephalus due to platybasia-narrow base of the skull, (ii) compressive myelopathy due to spinal canal stenosis at
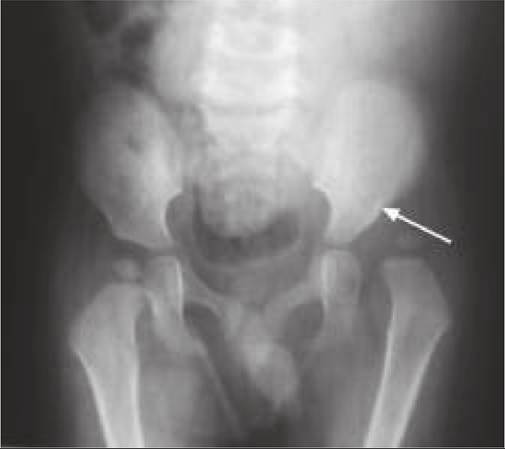
Fig. 23.7A and B: Achondroplasia (Clinical): (A) Appearance with short limbs; (B) Short vertebral pedicles with wedging.
cervical region (common in infants) or lumbar region (common in adolescents). Intelligence is normal, unless affected by hydrocephalus.
• Recurrent otitis media is common due to ear canal deformities, leading to conductive hearing loss in older children.
Diagnosis rests on clinical appearance simulating ‘circus dwarfs', anthropometry (short stature with higher US:LS ratio) and radiological evaluation. Important radiological signs include:
• Rhizomelic shortening of long bones with irregular, flared metaphyses (V-shaped or circumflex sign).
• Large calvarium with narrow base of skull and shorter facial bones
• Short vertebral pedicles with reducing inter-pedicular distance from L1 to L5, (in contrast to normal caudal widening). Lower thoracic or upper lumber vertebrae may show anterior tonguing/wedging and posterior scalloping (Fig. 23.7B).
• Short and broad iliac wings with horizontal acetabular roof.
Management aims to: (a) treat neurological complications, e.g. hydrocephalus or spinal canal stenosis, (b) correct orthopedic deformities, and (c) provide social and psychological support.
Prognosis: Life span is normal in absence of neurological complications. Post-marriage, risk of achondroplasia in offsprings is ~25% for having homozygous disease and ~50% for being heterozygous carriers. Prenatal diagnosis is possible for homozygous disease.
Hypochondroplasia is a milder variant of achondroplasia, where infant is stocky and muscular in appearance and short stature is not evident until late childhood. Craniofacial deformities are minimum except frontal bossing, and skeletal deformities are limited to some bowing of legs. Some cases have FGFR3 gene mutation at codon 540.
Thanatophoric dwarfism is the most severe skeletal dysplasia with extremely short limbs, small and pearshaped chest and severe radiological changes at birth with clover-leaf skull deformity (Kleeblatts chddel skull) in some cases. Most cases are stillbirths or die in neonatal period due to respiratory distress, owing to thoracic deformity. Some cases have FGFR3 gene mutation at codon 650.
Spondyloepiphyseal dysplasia (SED) is a heterogeneous group of uncommon chondrodysplasias, all characterized by short-trunk dwarfism due to abnormal development of vertebral bodies.
This group includes many disorders of variable severity, ranging from lethal variants in neonatal period, e.g. achondrogenesis (type II) or hypochondrogenesis, to milder variants, e.g. SED congenita, SED tarda or Kneist dysplasia.SED congenita, prototype of this group, manifests at birth or in infancy, with short trunk and neck, barrel shaped chest, relative rhizomelia, hypotonia with waddling gait and flattened vertebrae with delayed ossification on spinal X-rays. Disproportionate short stature worsens with age and many cases die in childhood due to respiratory compromise or develop neurological complications due to spinal deformities.
Osteogenesis imperfecta (OI) or Brittle-bone disease, is the commonest osteodysplasia (~1:20,000), characterized by a genetic defect in skeletal modelling with generalized decrease in bone density.
Etiology: OI is an autosomal dominant disorder, with primary defect in synthesis of type I collagen-main component of the extracellular matrix of bone and skin. Clinical spectrum: Easy fracturability of bones with consequent bony deformities is the hallmark of OI, though severity may vary from early lethal forms to very mild disease, as follows (Sillence classification):
Type I (mild) is characterized by a triad of: (a) recurrent fractures, (b) blue sclera and (c) early deafness in ~30- 60%. Similar family history is often present. Fractures begin in infancy or early childhood (not in utero, d/d other types), usually after trivial trauma and frequency reduces after puberty. Dental abnormalities, easy skin bruising and joint laxity is also common.
Type II (perinatally lethal) presents with multiple in utero fractures with severe deformities at birth, e.g. bowing of extremities, small beaded thorax due to rib fractures, large head with wide fontanels and dark blue sclera. Most cases are stillborn or die in early infancy.
Type III (progressive deforming) presents with severe and progressive deformities after repeated postnatal fractures since birth, though intrauterine fractures may occur.
Short stature, scoliosis with spinal compression and chest deformities are common. Sclera may be blue to white.Type IV (moderately severe) is an intermediate variant between type III and I as in utero fractures may occur (d/d type I) but postnatal fractures are more common after ambulation (d/d type III). Moderate bowing of long
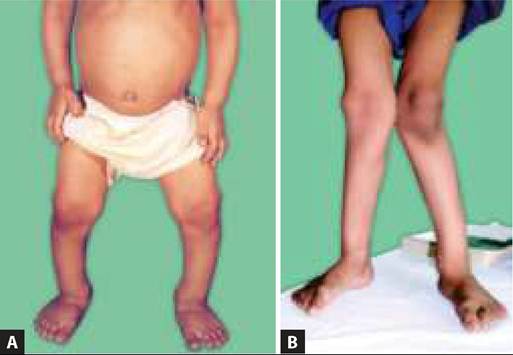
Fig. 23.8: Osteogenesis imperfecta: Multiple fractures.
bones with short stature is prominent feature in these cases, and fracture rate reduces after puberty. Sclera may be blue or white.
Type V and VI are rare cases similar to type IV disease but with different histology. A few cases of autosomal recessive type VII and VIII have been also reported.
Both, type I and IV disease, are also sub-classified as A and B, depending on the absence or presence of dental abnormalities (Dentinogenesis imperfecta), respectively.
Diagnosis must be suspected in any child with recurrent non-pathogenic fractures (Fig. 23.8) and confirmed by molecular studies or collagen biochemistry on skin biopsy (reduced type I collagen with altered type III:I collagen ratio on protein electrophoresis). Prenatal dia gnosis is possible in cases with family history on chorionic villous biopsy as early as at 12 week. Presence of intraunterine fractures on USG may be detected as early as at 16th weeks, suggesting severe disease.
D/D of OI includes other causes of recurrent pathological fractures, e.g. battered child, hemolytic anemia, osteoporosis/penia, hypophosphatasia, etc.
Management is non-specific and aims to reduce the frequency of fractures and deformities by: (a) avoidance of strenuous activities or sports, (b) early treatment of fractures, (c) surgical correction of deformities,
(d) orthotic support and physiotherapy.
Drugs, e.g. calcium or fluoride supplements are not effective, though growth hormone therapy or monthly IV biophosphonate infusions have been found of some benefit. Role of bone marrow transplant in severe cases is experimental.
Prognosis: Type I and IV may have normal life span but with disability, while Type II and III die in infancy or early childhood, respectively.
Common complications in survivors are: (a) functional deformities, (b) recurrent pneumonia due to thoracic deformities, (c) neurological complications, e.g. basilar invagination, hydrocephalus, etc. Recurrence risk in next sibling is 5-50% depending on the genotype.
Osteopetrosis (Marble-bone disease) is a rare but important disorder of defective bone resorption, leading
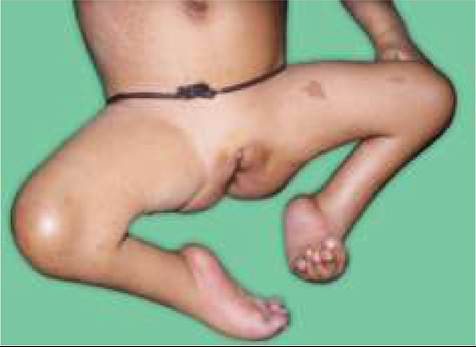
Fig. 23.9: Osteopetrosis: Bone in bone appearance.
to increased bone density and sclerosis with bone-in-bone appearance on X-rays (Fig. 23.9).
These cases present with: (a) failure to thrive, (b) severe anemia due to encroachment of marrow spaces, (c) hepatosplenomegaly and macrocephaly due to extramedullary erythropoiesis, (d) pathological fractures due to defective bone architecture, and (e) blindness, deafness or facial palsy, due to narrowing of cranial nerve exit-foramens.
More severely affected children have an autosomal recessive disease (usually TCIRG1 mutation) and die in infancy, while less severe autosomal dominant cases may survive till late childhood. Treatment is symptomatic though bone marrow transplant is useful in some cases.
23.5
More on the topic SKELETAL DYSPLASIA:
- Skeletal System
- Disorganized Cartilage and Fibrous Components
- CLINICAL SPECTRUM
- Defects of Tubular Bone or Spinal Growth Present at Birth
- CLASSIFICATION
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025