Sendai Virus Infection
Sendai virus was once common in laboratory rodent (mouse, rat, hamster, guinea pig) populations throughout the world, but is now rare. It is closely related antigenically to parainfluenza virus 1 of humans.
It is named after Sendai, Japan, where it was first isolated from laboratory mice inoculated with human lung suspensions and later isolated from naturally infected mice. Because of Sendai virus' close relationship with human parainfluenza 1 virus, there has been a long-held debate as to the human or mouse origin of Sendai virus, or if humans are naturally susceptible to Sendai viral infection. Studies have indicated that Sendai virus and human parainfluenza virus 1 replicate equally well in the upper and lower respiratory tracts of both African green monkeys and chimpanzees, suggesting that Sendai virus lacks a significant host range restriction and could very well be an anthropozoonotic agent. Sendai virus stands out among other murine viruses as an agent that is capable of causing significant clinical illness in adult, immunocompetent mice. Sendai infections may alter the prevalence of pulmonary neoplasms in experimental carcinogenesis studies. The squamous metaplasia that is found in recovering lungs has been misconstrued as neoplasia. Sendai virus is also known to perturb a variety of immune responses.Epizootiology and Pathogenesis
Sendai virus is a labile but highly contagious virus that is contact-transmitted by aerosol. Sendai virus prevalence appeared to increase during the 1960s and 1970s, when it was among the most common infectious disease agents of laboratory mouse populations, but has declined in the last few decades. Sendai viral epizootics had a peculiar seasonal pattern of unexplained origin. Although purely speculation, such a pattern could be explained if the virus were of human origin and circulating within the global human population.
Mice develop a descending infection of respiratory epithelium, which is abrogated by a cell-mediated immune response that clears the infection, but also generates disease. The level to which the infection extends within the respiratory tree is determined by mouse genotypic differences in mucociliary clearance, virus burden, and kinetics of immune response. Certain strains of mice, such as DBA/2, infant, and aged mice, are exquisitely susceptible to severe disease. These animals have an effective but delayed immune response to the virus, allowing infection to extend deep into the lung, but then mount a zealous response that results in severe disease. Other mouse strains, such as B6 mice, often have subclinical infections because of their rapid immune response, which precludes lower respiratory tract infection. In a comparative study among 19 inbred strains and 4 outbred Swiss stocks of mice, 129, DBA, and C3H mice were highly susceptible to lethal disease, whereas B6, AKR, SJL, and Swiss mice were resistant.
Sendai virus productively infects respiratory epithelium in the nose, trachea, bronchi, and bronchioles, as well as epithelium of the middle ears. It also spreads to type 2 alveolar cells. Cellular receptors for Sendai virus are widely distributed in many tissues, but respiratory tropism is dictated by the dependence of the virus to bud apically from respiratory epithelium and depend upon respiratory proteases to cleave its fusion glycoprotein into a biologically active form. Without the proteases, virus replication is restricted to a single cycle. Mutant strains of virus, with altered protease specificity and the ability to bud from basolateral cell membranes, can cause disseminated disease, but these (so far) are experimentally selected mutants. Following intranasal infection, virus titers appear in lung within a few days, then peak around days 6-8, at which time they rapidly decline due to the adaptive immune response. A transient viremia is present, reflecting the peak of virus activity in the lung.
In the preimmune phase, Sendai virus is only mildly cytopathic. As the acquired immune response mounts, there is infiltration of CD4 and CD8 T cells, resulting in CD8-triggered apoptosis of infected nasal, tracheal, bronchial, and bronchiolar epithelial cells with nonuniform exfoliation of epithelium and erosion. If infection has extended to the lower respiratory tract, there is interstitial alveolar inflammation. Infection is acute, with no persistent carrier state, except in immune-deficient mice.A number of innate and acquired components of the immune response are engaged during Sendai virus infection. Detectable seroconversion to Sendai virus appears around 10-12 days after infection and coincides with onset of immune-mediated clinical disease. Thus, in the preimmune phase of infection, mice may be mildly ill, but acute disease and mortality are associated with cytotoxic T-cell-mediated necrotizing bronchiolitis and alveolitis. Immunodeficient mice, particularly T-cell- deficient mice, develop progressive pneumonia. Maternal passive immunity is strongly protective for suckling pups when infection is enzootic within a population; it attenuates disease severity in postweaning mice that are exposed when passive antibody is waning.
Sendai virus infection is also associated with a number of infectious paraphenomena in mice and rats. It can predispose to the development of bacterial otitis media and interna, as well as precipitate Mycoplasma-associated lower respiratory disease in previously subclinically infected mice. Outbreaks of vestibular disease and pneumonia due to Mycoplasma or other bacteria can often be associated with recent activity of Sendai virus within the population. Although vertical transmission does not occur, Sendai viral infection of dams is associated with fetal resorption, prolonged gestation, and fetal death.
Pathology
Severely affected mice are dyspneic and have plumcolored consolidation of sharply demarcated foci, pulmonary hilus, anteroventral lung, or entire lung lobes (Fig.
1.35). These consolidated areas may turn gray in surviving mice. Microscopic changes during the immune phase of disease consist of segmental, necrotizing inflammation of nasal and airway epithelium (Fig. 1.36), as well as foci of interstitial pneumonia associated with terminal airways. Infiltrating cells vary with stage of infection, but include neutrophils, lymphocytes, and macrophages (Fig. 1.37). Alveolar spaces may be filled with fibrin, leukocytes, and necrotic cells, with atelectasis. Prior to immune-mediated necrosis, bronchiolar epithelium may be hypertrophic and hyperplastic, contain virus-induced syncytia, and may possess intracytoplasmic eosinophilic inclusions representing accumulation of viral nucleocapsid material. Intranuclear inclusions have been reported in nude mice. These virus-related changes are most apt to be seen in immature or immunologically deficient mice, since they are rapidly obscured by immune-mediated necrosis. During resolution, sloughed airway epithelium is replaced by proliferating hyperplastic epithelium, which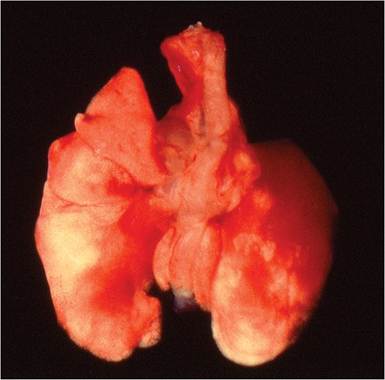
FIG. 1.35. Lung from a DBA mouse infected with Sendai virus. Note the dark areas around the hilus of both lungs consistent with congestion and early consolidation.
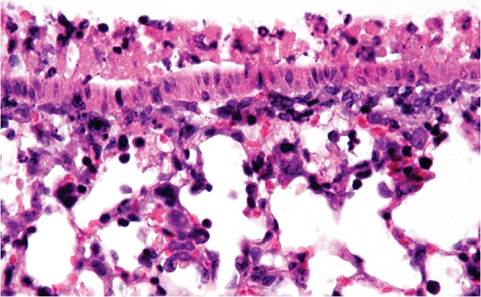
FIG. 1.36. Lung from a DBA mouse with Sendai viral infection. Note the acute necrotizing bronchiolitis.
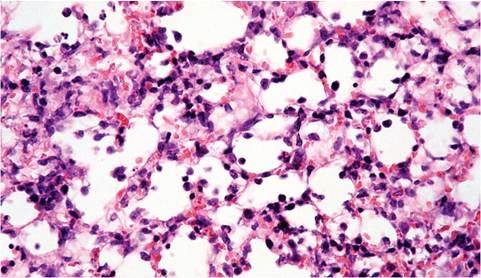
FIG. 1.37. Lung from a DBA mouse with Sendai virus infection, illustrating nonsuppurative interstitial pneumonitis.
may undergo transient but marked nonkeratinizing squamous metaplasia. Alveoli become lined by cuboidal epithelium (Fig. 1.38) or filled with metaplastic squamous epithelium. Increased lymphoid cells populate the bronchial tree, adventitia of adjacent blood vessels, and
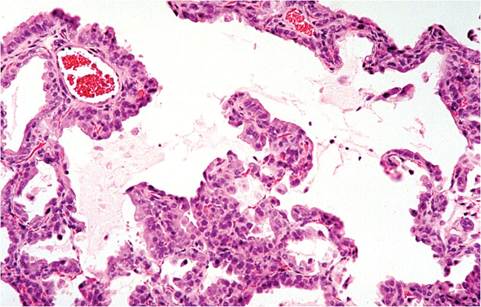
FIG.
1.38. Lung from a mouse in the reparative phase following recovery from Sendai virus infection. Note the cuboidal metaplasia of pneumocytes lining alveolar septa.
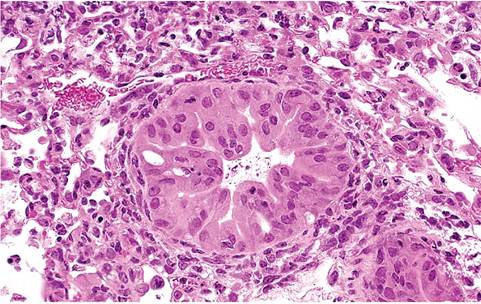
FIG. 1.39. Lung from a SCID mouse infected with Sendai virus.
There is marked hypertrophy and hyperplasia of bronchiolar epithelial cells that occur in the absence of immune-mediated necrosis.
alveolar septa. All of these changes completely resolve by the 3rd or 4th week. Severely affected lungs, in which progenitor alveolar cells have been destroyed, may have foci of fibrosing alveolitis and bronchiolitis obliterans. T-cell-immunodeficient mice develop progressive pulmonary consolidation with wasting. Their lungs tend to be pale, firm, and do not collapse. Because immunodeficient mice cannot mount an effective immune response, necrotizing changes are minimal, and airway epithelium is typically hypertrophic and hyperplastic (Fig. 1.39). These mice develop progressively severe, diffuse alveolitis, similar to progressive PVM pneumonia in immunodeficient mice.
Diagnosis
The clinical and microscopic features of Sendai viral pneumonia in immunocompetent adult mice are diagnostic and can be confirmed by seroconversion, which generally occurs coincidentally with clinical disease. Differential diagnoses include other causes of respiratory disease, such as Mycoplasma and Corynebacterium kut- scheri. Mild respiratory tract lesions can also occur with PVM or MHV. Immunodeficient mice can develop wasting disease with progressive pneumonia, which must be differentiated from pneumonia caused by PVM or P. murina. Sendai virus and PVM lesions in nude and SCID mice are similar, although in SCID mice, bronchial and bronchiolar lesions are more extensive with Sendai virus infection. Immunodeficient mice infected with PVM do not have the hypertrophic and hyperplastic changes in their respiratory epithelium that are characteristic of Sendai viral infections.