BASIC CONSIDERATIONS
Human genome: Each human somatic cell contains two copies of the entire genetic program, i.e. genome, each encoded in 3 million base pairs of DNA. However, less than 2% of human DNA is functionally active, rest being junk or redundant DNA, for which exact function are not yet known.
Most, but not all, of the DNA is contained in the nucleus, rest being in mitochondria or scattered in cytoplasm.DNA: DNA (deoxyribonucleic acid) is a double-stranded chemical structure, resembling a long and twisted rope ladder, i.e. helix. Each side of this ladder is made up of pentose sugar and phosphate molecules and each step comprises a pair of nitrogenous bases, termed base pairs (bp).
Human DNA contains four nitrogenous bases—two purines, i.e. Adenine (A) and Guanine (G); and two pyrimidines, i.e. Cytosine (C) and Thymine (T). Of these, A always pairs with T, and C with G (Fig. 11.1). The order of this bp arrangement varies from the site to site on a DNA strand, denoted as DNA sequence. DNA sequence at a specific DNA locus determines its role in genetic functions.
Chromosome: Entire DNA material in the somatic human cell nucleus is packaged in 23 pairs of chromosomes (n = 46), including 22 pairs of somatic chromosomes
Kshitija R Patil, Mukesh Agrawal
Fig. 11.1: Basic structure of DNA and chromosomes *C: cytosine; T: Thymine; A: Adenine; G: Guanine
(autosomes) and a pair of sex chromosomes (XX in females and XY in males). A baby receives one chromosome of each autosomal pair from different parents, one sex chromosome from father (X or Y) and another sex chromosome (X) from mother. Unlike somatic cells with paired chromosomes, (i.e. diploid cells), germ cells, e.g. sperms or eggs contain only 23 unpaired chromosomes, (i.e. haploid cells).
Morphologically, somatic chromosomes are of variable size and numbered according to its size in decreasing order-the largest being chromosome 1 and the smallest being chromosome 21. Sex chromosomes are not numbered, identified by their size and morphology. X is a large submetacentric chromosome, while Y is a small acrocentric chromosome.
Each chromosome consists of two identical thin strands (sister chromatids), with a short arm (p), long arm (q) and joined together by a centromere (Fig. 11.1). According to the location of centromere, chromosomes are also classified as metacentric (central centromere), submetacentric (centromere towards the short arm) and acrocentric (centromere at one end).
Gene: Gene is a functional unit of DNA, which codes for various proteins required for to govern various functions of the body. Each gene has a unique bp sequence, size, and location on a specific chromosome.
While over 1,00,000 genes are encoded in human DNA, all are not biologically active and some are active only in a particular cell, e.g. globin genes in RBCs. However, biologically inactive genes are not necessarily inert and some of them play important roles in modulation of the functions of biologically-active genes.
Genes with similar function, (e.g. globin chain synthesis) tend to cluster on a particular region of chromosome and may be switched-off and switched- on, at various stages of life. For example, #947;-globin gene, which determines synthesis of fetal Hb in utero is turned- off at birth, when #946;-globin gene is activated to produce adult Hb.
Each gene regulates synthesis of specific proteins and contains two functionally different regions, i.e. exons—which code its biological activity, and introns— which are either functionally inert or act as regulator of exons.
As one chromosome in each pair is derived from different parents, every gene on one chromosome has a counterpart gene at the identical location (homologous loci) on other chromosome of the pair, together termed as alleles.
Allelic genes are analogous, i.e. affect the same biological characteristics, but not necessarily identical. An individual is termed homozygous, if both alleles of a genetic pair carry identical biological information (normal or abnormal) and heterozygous, if they are nonidentical.Penetrance and expressivity: All genetic traits (genotype) do not necessarily manifest externally (phenotype) due to many factors including variable penetrance and expressivity of the Gene.
• Penetrance refers to the percentage of individuals with a specific genotype express in terms of corresponding phenotype, e.g. 80% penetrance means that 8 out of the 10 individuals carrying that particular gene manifest its effects.
• Expressivity refers to the degree that a particular genotype is expressed as phenotype in different persons, e.g. different severity of the disease despite same genetic defect.
Further, a gene may also not manifest due to presence of another dominant gene for same character (allele) on the other chromosome from different parental source, e.g. in autosomal or x-linked recessive inheritance, discussed later.
Transfer of genetic information for parental to daughter cells occurs during the cell division via transfer of DNA as chromosomes and constituent genes. Process of cell division may be divided into two types—meiosis and mitosis.
Meiosis is a two-step process that ultimately ends in formation of haploid germ cell. Subsequent fusion of two haploid germ cells from different parents restores normal diploid number in zygote, from which the new life begins.
Mitosis is characterized by replication of chromosomes before the cell division, so as to form two genetically identical, i.e. diploid daughter cells.
Mutation: All biological functions of a living cell are mediated by amino acids or proteins, e.g. enzymes. Genes are prime regulators of specific amino acid synthesis, essential for corresponding cell function.
Mutation (or variant), refers to a heritable change in the DNA sequence of the gene, which may or may not affect the structure and function of corresponding protein.
Mutations may be of three types: (a) point mutations or substitutions replacing single nucleotide from another; (b) deletions involving loss of one or more nucleotides in the gene; and (c) insertions, i.e. addition of one or more nucleotide in the gene. Each mutation may be benign, pathogenic or of unknown significance, based on the effect on phenotype. While germline mutations usually cause monogenic disorders, somatic cell mutations may cause cancers.Sequence variations without any obvious diseasecausing effect are termed as Polymorphism.
Mutations may occur naturally as chance errors in DNA replication during chromosomal division or may be due to external teratogens, e.g. ionizing radiation or chemical exposures.
11.2 MODES OF GENETIC INHERITANCE
According to the mode of transmission and inheritance, Genetic disorders may be classified into various types, as follows (Table 11.1):
1. Chromosomal Disorders
Chromosomal abnormalities are either numerical or structural and may be inherited from parents or arise de novo during gametogenesis by following mechanisms:
TABLE 11.1: Classification of genetic disorders
• Chromosomal disorders
- Numerical abnormalities
- Structural abnormalities
• Single gene (Mendelian) disorders
- Autosomal recessive or dominant
- X-linked recessive or dominant
- Y-linked inheritance (non-pathogenic)
• Multifactorial or polygenic disorders
• Mitochondrial disorders
• Alternate modes of inheritance
- Genomic imprinting
- Uniparental disomy
- Mosaicism
• Somatic cell disorders
• Meiotic nondisjunction, i.e. failure of a pair of chromosome to separate during meiosis. Consequently, one germ cell contains both members of chromosome pair (trisomy), while other lacks both (monosomy). Advanced maternal age is a major risk factor for meiotic nondisjunction.
• Anaphase lag, i.e. failure of one chromosome of a pair to move towards the opposite pole of dividing cell during anaphase of cell division despite separation, is another important source of numerical chromosomal abnormalities.
• Mitotic nondisjunction (mosaicism), i.e. failure of the separation of a pair of chromosome in diploid zygote, during initial cell divisions after fertilization. Consequently some of the individual cells are trisomic, while rest are monosomic. Mosaic defects tend to be less severe or sub-clinical.
Numerical chromosomal abnormalities include the number of chromosomes being other than the haploid number (23) of germ cells or diploid number (46) of somatic cells. Aneuploidy, the commonest numerical abnormality, refers to number of chromosomes being other than in multiples of haploidy, due to more chromosome/s (47XY/XX or Down syndrome) or less chromosome/s (44XO, Turner syndrome). Other numerical abnormalities include Euploidy and Polyploidy (Table 11.2).
Structural chromosomal abnormalities include (a) Deletion, i.e. loss of a DNA segment, (b) Translocation, i.e. separation of a DNA segment from one chromosome and re-attachment on another chromosome, and (c) Inversion, i.e. separation of a DNA segment from one chromosome and its re-attachment on the same
TABLE 11.2: Glossary of chromosomal abnormalities
Normal karyotypes
• Haploidy: Cells with 23 chromosomes, e.g., germ cells
• Diploidy: Cells with 46 chromosomes, e.g. somatic cells Numerical abnormalities
• Euploidy: Number of chromosomes in multiple of haploidy (46, 69,...)
• Polyploidy: Euploidy with chromosomal numbers gt; diploidy (69, 92,..)
• Aneuploidy: Number of chromosomes not in multiples of haploidy.
• Trisomy: Presence of extra chromosome (47 XY/XX, Down syndrome)
• Monosomy: Absence of one chromosome (45 XO, Turner syndrome)
Structural abnormalities*:
• Deletion: Loss of a chromosome segment (Cri du chat syndrome)
• Translocation: Inter-chromosome transfer of DNA segment
• Inversion: Detachment of DNA segment gt; re-attachment in reverse order
*also see Fig. 11.2
Fig.
11.2: Common structural abnormalities of chromosomes.chromosome but in reverse order. Mutual exchange of DNA segments between two chromosomes is termed as Reciprocal translocation. However, inversion or reciprocal translocation rarely manifest clinically, due to no loss of DNA segment (Fig. 11.2).
Robertsonian translocation is a specific translocation defect involving fusion of two acrocentric chromosomes near their centromere with loss of short arms, leading to reduction in total chromosomal numbers, (e.g. 45). Though the short arms of acrocentric chromosomes have no known function and carriers of Robertsonian translocations are phenotypically normal, they are at-risk for abortions.
Microdeletions, i.e. deletions, which are not visible on routine karyotyping and need special prophase studies for diagnosis, are being increasing recognized in association with various genetic disorders, e.g. Prader- Willi syndrome, William syndrome, Alagille syndrome, etc. Some important chromosomal disorders have been discussed in Ch 11.3.
Loss or gain of smaller regions of a chromosome, known as copy number variations (CNV), usually involve more than one gene and are implicated in many human diseases.
2. Single Gene (Monogenic) Disorders
As stated earlier, each gene has an analogous allele at the identical location on other chromosome of the pair affecting same biological characteristics, but may or may not be identical (homozygous vs heterozygous).
In heterozygous state, generally one gene of the allelic pair determines biological activity (dominant gene), while other remains suppressed (recessive gene) and manifests only in absence of dominant allele. Thus, disorders encoded on dominant genes manifest even in a heterozygous state, while disorders of recessive genes
are asymptomatic in heterozygous state (suppressed by normal allele) and manifests only in homozygous state.
As the genes present on X-chromosome of a male (XY) do not have their corresponding allele (hemizygous state), an X-linked recessive gene manifests only in males and remains suppressed in heterozygous females (XX).
Thus, single gene disorders may be broadly divided into 4 categories—(a) autosomal dominant, (b) autosomal recessive, (c) X-linked recessive, and (d) X-linked dominant disorders. Y-linked genetic abnormalities are rare and present with minor phenotype features in males, inherited from fathers, e.g. hairy pinna, frontal baldness, webbed toes, porcupine skin, etc.
Autosomal dominant (AD) disorders, i.e. defects encoded on the dominant gene on autosomal chromosome, are characterized by following features (Fig. 11.3):
• Clinical manifestation in heterozygous state, as the defect is encoded on the dominant gene of the pair. Homozygous states are rare for ADDs and present with more severe disease.
• No silent carriers, as all genetically abnormal members of the family manifest clinically, unless the gene has not manifested due to incomplete penetrance.
• Vertical inheritance, i.e. history of similar disease in one of the parents (or previous generations), unless the genetic error is a new de novo mutation.
• Usual risk of recurrence during each pregnancy being 50%, when one parent is heterozygous and another is normal. In rare cases, it may be even higher if one parent is homozygous (100%) or both parents are heterozygous (75%).
• Both sexes are equally affected, as the defective gene is present on the autosomes.
• Most ADDs are non-lethal and hence, more common than recessive defects due to survival of affected individuals till reproductive age.
Common examples of ADD are achondroplasia, neurofibromatosis, Marfan syndrome, hereditary spherocytosis, etc.
Autosomal recessive (AR) disorders, i.e. defects encoded on the recessive gene of autosomal chromosome, are characterized by following features (Fig. 11.4).
• Clinical manifestations only in homozygous state, due to absence of corresponding dominant gene to suppress mutant gene activity.
• Silent carrier state in heterozygous individuals, due to suppression of abnormal gene by corresponding dominant gene. However, some carriers may have borderline abnormalities.
• Horizontal inheritance, i.e. all members of same generation are genetically affected, when a heterozygous carrier mates with another heterozygous carrier. However, some of them manifest with disease (if homozygous), while rest become carriers (if heterozygous). Parental history of similar disease is absent in these cases, who are heterozygous carriers.
• Usual risk of recurrence in subsequent pregnancy is 25% for homozygous disease and 50% for heterozygous carriers, if both parents are carriers. All children of a homozygous patient (very rare) are carriers, if the other partner is normal.
• Consanguinity is an important risk factor for ARDs, as the members of same family tree are more likely to be heterozygous carriers, than the general population.
• Both sexes are equally affected, as the defective gene is present on the autosomes.
• ARDs are frequently lethal and hence, relatively rare due to unlikely survival of a diseased case till reproductive age.
Common examples of ARDs are severe forms of inborn errors of metabolisms, hemoglobinopathies and skeletal dysplasias, etc.
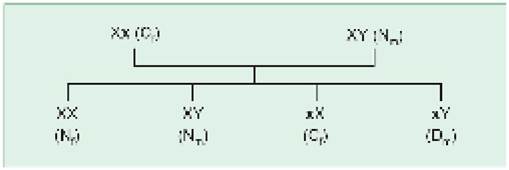
X-linked recessive (XLR) disorders denote a defect of recessive gene located on X chromosome, characterized by following features (Fig. 11.5):
• Clinical manifestations only in males (XY), due to absence of corresponding dominant gene to suppress the abnormal recessive gene. Females (XX) are not affected due to presence of normal dominant gene, except in very rare instances of 45, X genotype (Turner syndrome), X autosome translocation, skewed X inactivation, fresh mutations or a diseased male mating a carrier female. However, carrier females may have mild and silent biochemical defects, e.g. low factor VIII activity in hemophilia.
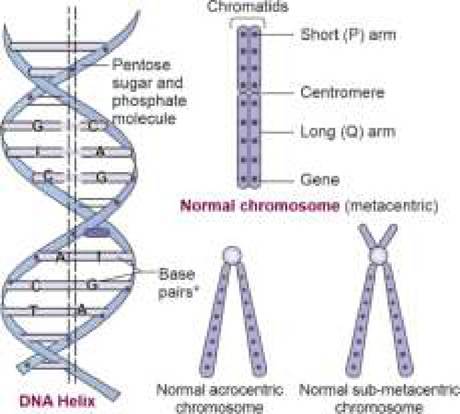
Fig. 11.3: Autosomal dominant inheritance.
Bold ‘A' denotes abnormal dominant gene
N: normal, D: diseased
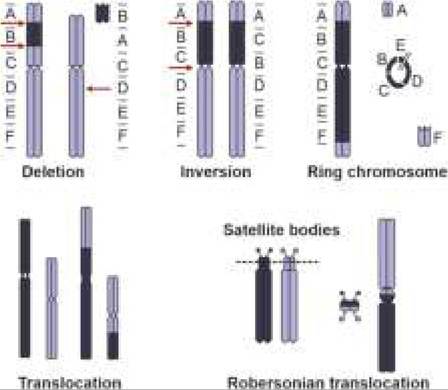
Fig. 11.4: Autosomal recessive inheritance.
Bold ‘a' denotes abnormal recessive gene N: normal; D: diseased; C: carrier
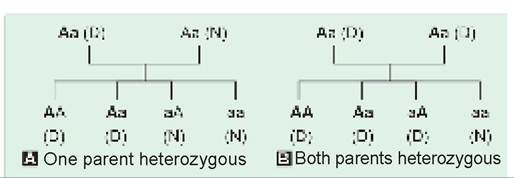
Fig. 11.5: X-linked recessive inheritance.
N: normal, D: diseased, C: carrier
Genes in bold letters denote abnormal gene, subscripted letters m and f denote male and female.
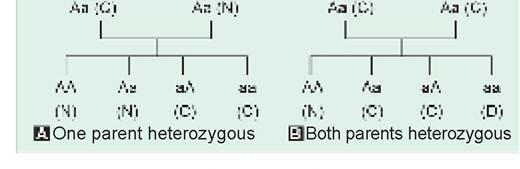
Fig. 11.6: X-linked dominant inheritance.
N: normal; D: diseased
Genes in bold letters denote abnormal gene, subscripted letters m and f denote male and female.
• Carrier state only in females (XX), as males are always affected by disease.
• Oblique transmission, i.e. only males on maternal side, (e.g. maternal uncles) are affected with disease, while mother is an asymptomatic carrier and father is normal.
• Usual risk of recurrence during next pregnancy is 50% for disease in males or carrier state in females, when mother is a carrier.
Common examples of XLRs are hemophilia, Duchenne muscular dystrophy, color blindness, G6PD deficiency, etc.
X-linked dominant (XDD) disorders are uncommon and characterized by (Fig. 11.6):
• Clinical manifestations in both sexes, as females are also affected due to dominant nature of abnormal gene.
• Risk of recurrence is 50% if mother in heterozygous and father is normal. However, all female offsprings of a hemizygous father are affected.
• Disease tends to be less severe in females? due to modifying effect of corresponding normal allele, thus creating excess of females in XLD families.
• As males are more severely affected, defective gene is usually acquired from maternal side.
Common examples of XLDs are familial hypo- phosphatemic rickets, incontinentia pigmenti, orofacial digital syndrome, nephrogenic diabetes, etc.
3. Polygenic and Multifactorial Disorders
Often confused together, these two terms have different connotations. While polygenic disorders represent a quot;defect regulated by additive or interactive effect of two or more genesquot;, multifactorial disorders refer to a quot;genetically determined susceptibility of certain individuals to various environmental factors”. Various normal human traits, e.g. height, as well as abnormal disorders are multifactorial in origin. In these cases, contribution of genes is termed heritability.
While exact genetic error in most MFDs is usually not clear, some have strong association with certain specific HLA types, e.g. diabetes (DR3/4), spondyloarthropathy (B27), etc.
TABLE 11.3: Recurrence risk in multifactorial disorders
*If previous sibling is affected, with normal parents.
Risk of recurrence (RR) in MFDs, in absence of well- defined genetic basis, is usually estimated on the basis of epidemiological observations (Table 11.3). Some general observations are as follows:
• RR in most MFDs is ~2-10% among first-degree relatives, and gradually declines in more distant relatives.
• RR rises by 2-3 folds, when more than one family members, e.g. one parent and one sibling or two siblings are affected (d/d single gene disorders, where it is same for each pregnancy).
• RR is higher in common and severe disorders, as compared to rare or benign defects.
• In disorders with sex-predilection, RR is higher if the index case belongs to rarely affected sex, e.g. in Infantile hypertrophic pyloric stenosis, which is common in males, RR is higher for offspring of an affected mother than of affected father.
• Unlike single gene defects, presence of similar MFD disorder in identical twins (concordance) is never 100%, usually being 21-63%.
4. Mitochondrial Disorders
Mitochondria contain ~16,000 bps of DNA, arranged in small circular segments (mitochondrial chromosomes). Mutations in mitochondrial genome are responsible for many genetic disorders, e.g. Leber hereditary optic neuropathy, MELAS (Mitochondrial myopathy
Encephalopathy, Lactic acidosis and Stroke), Leigh syndrome and MERRF (myoclonic epilepsy with ragged- red fibers). Mitochondrial disorders are characterized by:
• Involvement of specific tissues, e.g. brain, muscles and eyes.
• Exclusive inheritance from mothers, as sperms do not contain mitochondrial component. However unlike X-linked disorders, offsprings of both sexes are affected in these disorders.
3. Non-traditional Modes of Inheritance
Non-traditional modes of inheritance include some genetic traits, which are not determined by an equal genetic contribution from both parents or do not follow conventional modes of inheritance discussed earlier. Some important disorders of this nature are as follows: Uniparental disomy (UPD) denotes a mode of inheritance in which both chromosomes (or part of a chromosome) of a pair are inherited from same parent. Most important effect of UPD may be increased risk of manifesting autosomal recessive disorders, due to transfer of two identical chromosomes carrying mutant genes from a heterozygous parent. UPD has been observed in many recessive disorders, e.g. cystic fibrosis or other defects,
e. g. Prader-Willi syndrome and Russell-Silver syndrome.
Genomic imprinting refers to the differences in the phenotype expression of a genetic defect, depending on the parent of origin for implicated gene/chromosome. For example, deletion of paternally-derived 15th chromosome presents with Prader-Willi syndrome, while similar deletion of maternal-derived chromosome manifests as Angelman syndrome.
Mosaicism refers to an individual with two different cell lines, derived from a single zygote (mitotic nondisjunction). Over 2% of conceptions are estimated to have mosaic chromosomal abnormality in first trimester. Mosaic defects tend to be less severe and often allow the survival in otherwise lethal chromosomal defects due to presence of normal cell lines. Mosaicism may be divided into:
• Chromosomal mosaicism, e.g. Down syndrome (46/47), Turner syndrome (45,X/46XX);
• Single gene mosaicism, limited to certain genes, e.g. McCune-Albright syndrome, and
• Germ-line mosaicism, i.e. presence of mosaicism in germ cells. Unlike other types, germ-line mosaicism is inherited from parents and hence, may affect more than one offspring of mosaic parents, e.g. in Duchenne muscular dystrophy or osteogenesis imperfecta.
Chimerism refers to an individual with at least two or more cell lines, often originating from fusion of many different zygotes (d/d Mosaicism with single zygote).
4. Somatic Cell Disorders
The term ‘somatic cell disorders' refers to a new genetic mutation during mitotic cell division in the somatic cells, after differentiation of gonads. These mutations are limited to a single cell line and not transmitted to the next generation. Although each individual is likely to have many somatic mutations, most of them are silent and never detected. Two important examples of somatic mutations are cancers due to mutation in oncogenes, and autoimmune disorders due to mutation in genes regulating T-cell selection.
11.3 CHROMOSOMAL DISORDERS
Chromosomal abnormalities are present in ~0.4% of live births and ~50-60% of aborted fetus and include numerical as well as structural abnormalities. These abnormalities may be inherited from parents or arise de novo during gametogenesis.
When to suspect? Unlike singe gene disorders, Chromosomal disorders do not follow a specific pattern of inheritance and hence, family history is of limited significance in diagnosis. Physical dysmorphism is the hallmark of most chromosomal disorders, though Table 11.4 enlists some common indicators of chromosomal disorders. Diagnosis is often based on constellation of clinical features many good databases are available to assist in the search for diagnostic possibilities.
Laboratory diagnosis of chromosomal disorders includes study of chromosomal number and morphology, using following methods:
• Conventional karyotyping may be performed on any living cells, e.g. lymphocytes, biopsies, aspirates, etc. or even the chorionic villi and amniotic fluid for prenatal diagnosis. Since the chromosomal profile is best appreciable during metaphase of mitotic division, karyotyping involves cell cultures to stimulate the mitotic division, arrest the division in metaphase by adding colchicine. dispersing these multiplying chromosomes by bathing the sample in hypotonic solution and then Staining with conventional stains, e.g. G-banding (Giemsa stain). In these stained slides, each chromosome is identified under microscope due to its unique light and dark banding patterns.
TABLE 11.4: Clinical indicators of chromosomal disorders
• Family history of...
- Similar morphology/disease with unexplained etiology
- Recurrent fetal or neonatal deaths or malformations
• Physical characteristics
- External dysmorphic features
- Multiple congenital malformations
- Developmental delay/Intellectual disability
- Ambiguous genitalia or sexual infantilism
- Abnormal stature or deformities
- Unusual cancers
• Prenatal biochemical/USG abnormalities
- Abnormal USG
- Abnormal biochemical markers
Conventional karyotyping require long time (2-3 weeks) for results and can identify numerical and structural chromosomal abnormalities, but only up to 5 Mb resolution.
• Molecular cytogenetic studies, e.g. Fluorescent in situ hybridization (FISH), quantitative Polymerase chain reaction (qPCR) and Multiplex ligation dependent probe amplification (MLPA) use target-specific fluorescent probes or markers against the suspected chromosome/s or regions. Results are available rapidly within 24 hours but only for the defect/s in that specific chromosome/s or regions. These tests are very useful where clinical suspicion limits the diagnostic possibilities, e.g. identification of aneuploidies for prenatal diagnosis and microdeletions.
• Chromosomal microarray has revolutionized the diagnosis of chromosomal disorders, based on the principle of complementary DNA hybridization to screen entire genome for differences from the known pattern, termed as copy number variations (CNVs), i.e. deletions or duplications. This test does not require living cell, results are available in few days and defects can be identified with a fine resolution of as low as 10-20 kb. However, microarray does not detect balanced translocations which do not alter CNVs and point mutations, deletions or duplications at the single gene level.
Chromosomal microarray is useful when there is a need to screen for many defects due to lack of a specific genetic diagnosis, e.g. unexplained intellectual disability, autistic spectrum disorders and multiple congenital anomalies.
Barr body (BB): In females, one of the two X chromosomes is randomly inactivated during early embryogenesis (Lyon hypothesis), which may be seen as a clump of heterochromatin adjacent to the nuclear membrane (termed Barr body). Males have only one X chromosome and hence no Barr body. Thus, the number of BB is one less than the number of X chromosomes. Detection of a Barr body in buccal smear is a quick method to identify the genetic sex in ambiguous genitalia (Female: one BB, Male: no BB) or disorders with abnormal X numbers, e.g. Turner syndrome (45 XO; no BB), 47 XXY female (2 BB), etc. However, these results are often misleading and no longer used in practice.
Some common chromosomal disorders are discussed as follows:
11.3.1
More on the topic BASIC CONSIDERATIONS:
- Agrawal M.. Textbook of Pediatrics. 3rd ed. — CBS Publishers,2025. — 973 p., 2025
- EMPIRICAL MODELS
- Developments in the New Welfare Economics and the Economic Theories of Justice
- Sources of Contemporary Institutionalist and Evolutionary Theory: Four Unconventional Economists
- North Korea's Cultural Revolution in 1972
- Problems of Economic Dynamics
- FIVE COMPONENTS OF LEGAL COMPETENCIES
- Background Context
- REVIEW OF FORENSIC ASSESSMENT INSTRUMENTS
- I ABNORMAL GENITAL BLEEDING ^461 ^593