DOWN SYNDROME
Down syndrome (DS) is the commonest chromosomal aneuploidy in live-births, characterized by a typical phenotype and triplicate copy of entire chromosome 21 or its critical segment (DCR).
Incidence of DS in general population is ~1:600-800 live births, though the true incidence is much higher as ~50% of affected conceptions are spontaneously aborted. Advanced maternal age is the most important risk factor for DS, with an estimated risk of ~1:1000 below 30 years, ~1:350 by 35 years and ~1:100 after 40 years.
Cytogenetically, DS may be divided into two types-
(a) regular trisomy (~95%), i.e. presence of entire extra chromosome (47,XX#8725;XY,+21); (b) translocations (~5%), i.e. presence of 3 copies of DCR, but with normal chromosomal number (46, XX#8725;XY).
• Regular trisomy is the commonest cause of DS in children born to elderly mothers, due to nondisjunction of 21st chromosome during meiotic phase in 94% cases or during mitotic phase in 1%. In 95% cases of regular trisomy, the extra chromosome is derived from mother.
Non-disjunction in mitotic phase leads to mosaicism, in lt;1% cases witth presence of two different cell lines, one normal and another with trisomy.
• Translocations, i.e. the transfer of additional DCR by transfer on other acrocentric chromosomes (Robertsonian translocation), e.g. 14, 15, or 13 (usually t14;21), may arise de novo or inherited from a balanced carrier parent. Most are maternally inherited and only 20% are paternal. Maternal age is irrelevant and most cases of DS born to younger mothers are translocations.
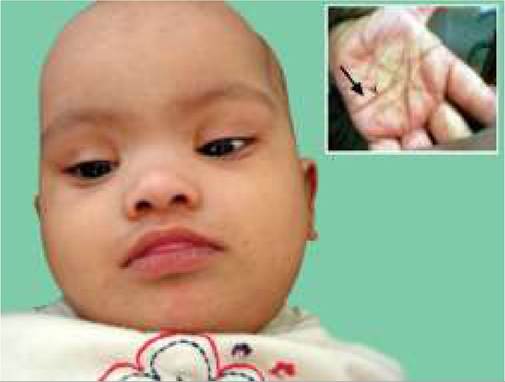
Clinical phenotype of DS spans from presence of a few dysmorphic features to full-blown picture including developmental delay, hypotonia and systemic defects (Fig. 11.7).
Common systemic problems present since birth include congenital heart diseases in ~40% (commonest being atrioventricular septal defect), gastrointestinal malformation, e.g.
duodenal atresia in ~12%, hypothyroidism in ~15-50% and hearing defects in ~40-60%, while some others may arise after few years, e.g. atlantoaxial joint instability in ~10-30% and leukemia or other lymphoprolierative disorders in later life (Table 11.5).
Fig. 11.7: Down syndrome: Facial appearance and simian crease (Inset).
TABLE 11.5: Clinical features in Down syndrome
• External dysmorphism
- Head: Brachycephaly or hat occiput, microcephaly, Delayed closure of fontanels
- Eyes: Upward slanting palpebral fissures, hypertelorism, Epicanthic folds, brushfield spots, cataract, refractive errors
- Nose: Small and depressed nasal bridge, fiat face
- Ears: Small/dysplastic ears, deafness
- Oral cavity: Dental hypoplasia, furrowed protruding tongue, high-arched palate
- Neck: Short neck** with loose posterior folds in infancy
- Hands/feet: Short/broad hands, clinodactyly**, sandal
- gap (#8593; gap between 1st and 2nd toe)
- Dermatoglyphics: Simian crease** and others*, plantar (Kennedy) crease between 1st and 2nd toe
- Skin: Dry, hyperkeratotic skin, fine sparse hairs, Cutis marmorata
- Genitals: Cryptorchidism, micropenis
• Generalized hypotonia with hyperextensible joints hypotonia**, poor Moro's reflex**, hyper-extensible joints**
• Developmental delay
• Systemic abnormalities
- CVS: Endocardial cushion defects, other CHDs
- GIT: Obstructive lesions, e.g. duodenal atresia, Imperforate anus, Hirschsprung disease
- Respiratory: Obstructive sleep apnea syndrome, hypoplastic lung, pulmonary hypertension
- Endocrinal: Cong hypothyroidism, short stature, Infertility
- Hematological: Leukemia, leukamoid reactions
- Skeletal: Hypoplastic pelvis**, atlanto-axial instability
- Others: Visual errors, deafness
*wide ‘ATD' angle, distal triradius, ulnar loop ridge patterns **included in Halls criteria for Down syndrome
Despite developmental delay, these babies are friendly, playful and enjoy music.
Motor development is slower than social development due to hypotonia, which tends to improve with age.While many cases survive till adulthood and lead relatively independent life after learning the self-help skills, ~50% die in early childhood or need recurrent hospitalizations due to congenital heart diseases and recurrent aspiration (palatopharyngeal incoordination). Diagnosis is largely clinical, though karyotyping is indicated in all cases to confirm the diagnosis, mode of inheritance and genetic counseling.
Management aims to: (a) improve the quality of life,
(b) detect and treat systemic conditions, and (c) genetic counseling; and includes:
• Confirmation of diagnosis by karyotyping. If it is due to translocation, cytogenetic studies of parents are also necessary to detect balanced carriers and assess the risk for next conception.
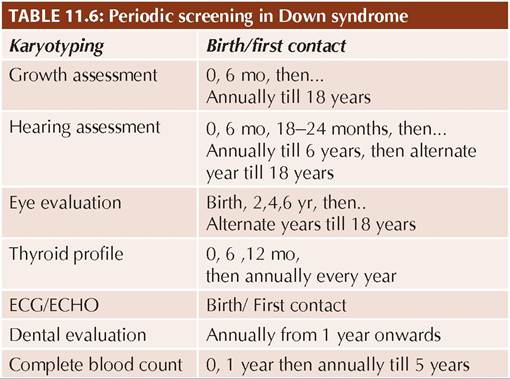
• Screeningfor systemic abnormalities, e.g. congenital heart diseases, hypothyroidism and hearing/vision defects is essential at birth and then periodically, as many defects become apparent only in later life. IAP has
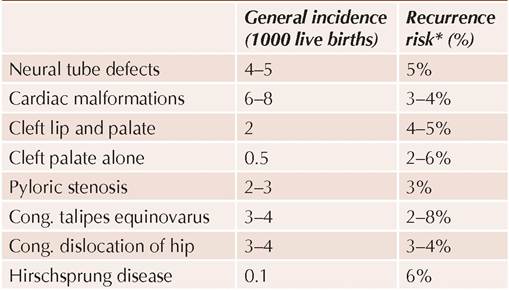
TABLE 11.7: Recurrence risk for Down syndrome
recommended a time schedule for periodic follow-up for important medical issues till 18 years of age in all cases of DS (Table 11.6).
• General care to ensure adequate nutrition, immunization, and growth and development monitoring.
• Interventional programs, e.g. physiotherapy, occupational therapy and self-help learning to achieve maximum functional independence.
• Psychological support to family.
• Medical therapies, i.e. megavitamins, selenium, piracetam, dimethyl sulfoxide, etc. have been tried in DS but with no proven value. However, associated illnesses, e.g. hypothyroidism should be promptly treated. Surgical intervention is indicated for major structural malformations.
Cosmetic surgery for facial dysmorphism is justifiable in selected cases to increase social acceptance.Genetic counseling is the most important component of management in DS and involves:
• Assessment of recurrence risk in future pregnancy: Recurrence risk for Down syndrome varies with maternal age and parental cytogenetic profile (Table
11.7).
• Prenatal screening based on biochemical and USG markers (Table 11.8) is indicated in high-risk mothers, e.g. advanced age, previous affected child and abnormal parental karyotype, as follows:
- First trimester screening (11-13 weeks) usually rests on: (i) reduced Pregnancy associated plasma
TABLE 11.8: Prenatal screening of Down syndrome
protein-A (PAPP-A) levels, (ii) elevated human chorionic gonadotropin (HCG) levels and (iii) increased nuchal thickness gt; 4 mm (echo-free space between skin and dorsal cervical spine).
Non-invasive prenatal screening (NIPS) is a new screening test in high-risk cases to analyse free fetal DNA in maternal plasma at ~10th week of gestation. It can detect aneuploidies of chromosome 21 with 99% sensitivity but false-positive results are possible and must be confirmed with cytogenetic studies.
- Second trimester screening (14-16 weeks) involves triple/quadruple marker test and includes presence of a combination of biochemical abnormalities in mother: (i) reduced-fetoproteins levels, (ii) reduced unconjugated estradiol levels, (iii) elevated human chorionic gonadotropin levels and (iv) increased inhibin levels in Quadruple marker test. A positive test indicates gt;50% risk of DS. Presence of certain USG abnormalities, e.g. heart disease or duodenal atresia also indicate need to exclude DS.
Important prenatal USG markers of DS include (Two or more of following): (a) increased nuchal translucency, (b) absent or hypoplastic nasal bone, (c) presence of soft markers like pyelectasis, echogenic bowel, echogenic intracardiac focus, single umbilical artery, choroid plexus cyst, short femur, etc., (d) structural abnormalities, e.g.
duodenal atresia, cardiac defect, and (e) adjunct features like clinodactyly.• Confirmation of prenatal diagnosis, by cytogenetic studies on suitable antenatal samples is possible in high-risk cases as early as 11-13 weeks by chorionic villous sampling, 17-23 weeks by amniocentesis and 18 weeks on cordocentesis. While conventional karyotyping takes ~2 weeks, PCR-based tests are available to give results in 2-3 days.
• Parental counseling: DS is a shattering diagnosis for parents and they should be informed as soon as possible after the confirmation in simple terms about: (a) confirmation of diagnosis, (b) possibilities of associated problems in present and in future, (c) need for regular follow-up to detect and mange these problems, (d) expected level and pace of development and achievement of life-skills, (e) risk of recurrence and options for prenatal diagnosis during next conception.
They must also be counselled about immediate risks for life due to congenital anomalies. If any, morbidities which can arise in future and precautions required to minimize them. It needs to be emphasized that most cases of DS are trainable and can live near normal adult life with family support. Parents must be answered truthfully but compassionately for their apprehensions and may also be connected to support groups. Issues, e.g. schooling, menstrual hygiene etc may be discussed in future dates.
Other important somatic chromosomal disorders are as follows:
Trisomy 18 (Edward syndrome) is seen in ~1:6000 births, characterized by low birth weight, microcephaly, rocker-bottom feet, joint contractures/deformities overlapping fingers, developmental delay and cardiac/ renal malformations. 95% of these cases die in infancy.
Trisomy 13 (Patau syndrome) is seen in ~1:10,000 births, characterized by multiple midline defects, e.g. facial defects (hypotelorism, bulbous nose, cleft palate, deformed ears, microphthalmia), cerebral malformations (microcephaly, encephalocele or holoprosencephaly), chest deformities (hypoplastic ribs, congenital heart diseases) and genital abnormalities (hypospadias).
Polydactyly with overlapping fingers and visceral malformations are common and most cases die soon after birth.Cri-du-chat syndrome, i.e. deletion of short arm of 5th chromosome (5p-), is characterized by typical cat-like cry, hypotonia, microcephaly with moon-like facies and mental retardation.
Microdeletion syndrome are characterized by multiple and small chromosomal deletions, missed on routine karyotyping studies.
Many microdeletion syndromes are being identified with newer diagnostic methods, commonest being William syndrome (typical elfin facies with supravalvular aortic stenosis), Prader-Willi syndrome (obesity, short stature, hypotonia), Rubinstein-Taybi syndrome (microcephaly, beaked nose with low philtrum, broad and long thumbs/toes), DiGeorge syndrome (first brachial
arch defects, e.g. thymus aplasia, hypocalcemia due to hypoparathyroidism, ear defects and congenital heart diseases) and Alagille syndrome (intrahepatic biliary atresia).
Chromosomal breakage syndromes: Some chromosome sites are known to show abnormal tendency for separation, breakage or attenuation, either spontaneously or under environmental stress, e.g. UV radiations (Fragile sites). Many genetic disorders have been identified with fragile chromosomes, e.g. Fanconi anemia, Bloom syndrome (skin malignancies with mental retardation), ataxia telangiectasia, etc., as well as higher risk of malignancies.
11.3.2
More on the topic DOWN SYNDROME:
- REFERENCES
- Obesity and preconceptional counselling
- Age and fertility
- Screening for chromosomal abnormalities
- 12 Congenital Anomalies
- Reye’s-Like Syndrome
- REFERENCES
- Schwartz-Jampel Syndrome (Chondrodystrophic Myotonia)
- Diagnostic procedures
- Screening for Fetal Aneuploidy in Multiple Pregnancy