DIAGNOSTIC TECHNIQUES
Precipitin and Agglutination Tests
Two of the earliest serological tests used are the precipitin test and agglutination tests. The precipitin test is used with antigen particles too small to be seen with a light microscope, such as viral particles (Lancaster 1966, Hedger 1981, Seymour and Yuill 1981).
The antigen being used and serum being tested are combined in a capillary tube, on a slide, or in an agar-gel test (Ouchterlony 1958, Coleman et al. 1989). If the specific antibody is present, it combines with the antigen and the resulting lattice framework forms a milky precipitate (precipitin). The agglutination test uses the same theory, but is used with larger antigen particles such as bacteria (Damon and Johnson 1944, Thal 1956, Bell 1981, Witter 1981) and occasionally large viruses such as poxviruses (Ledingham 1937). Larger antigens make the lattice framework (agglutination) a little more evident and thus require less specific antibody for detectable positive tests, making them more sensitive.Passive Hemagglutination Test
Increasing antigen size to decrease the antibody concentration needed for detectable antigen-antibody reactions was enhanced further with the passive hemagglutination test (PHA). Here the test antigen is chemically attached to the relatively large structure of an erythrocyte; this antigen-erythrocyte combination is exposed to the serum from a test animal (Mahony and Chernesky 1999). Antibody against the test antigen causes a clumping of the carrier erythrocytes (hemagglutination). Since hemagglutination is relatively easy to detect, compared to the finer precipitates of the precipitin or agglutination tests, the PHA requires less specific antibody for detecting a positive reaction. The PHA is used to detect antibodies against a variety of bacteria (Wright and Feinberg 1952, Cavanaugh et al. 1965, Wetzler et al.
1968) and viruses (Hierholzer et al. 1969).Hemagglutination Inhibition Test
The hemagglutination inhibition (HAI) test is based on the observation that some viruses spontaneously agglutinate erythrocytes in vitro (Mahony and Chernesky 1999). For example, myxoviruses and paramyxoviruses were named because they are very attracted to the mucins (myxo: mucins) that coat erythrocytes. Some of the antibodies against these viruses specifically inhibit their hemagglutinating antigens. In the HAI test, the known viral antigen and unknown serum being tested are combined. If the antibody is present in serum, it combines with the virus and covers the hemagglutinating sites. Sheep erythrocytes are added. If the antibodies against the virus are present, there is no hemagglutination because the hemagglutinating antigens on the virus already are covered by specific antibody. If the specific antibody is lacking in the tested serum, the virus antigen sites still are exposed, and are able to cause hemagglutination of the sheep erythrocytes. Thus a positive test for the antibody is indicated by a lack of hemagglutination. This test is used for influenza and parainfluenza viruses (Vaananen et al. 1985), arboviruses (Seymour and Yuill 1981), and some other viruses (Hierholzer et al. 1969).
Neutralization Test
The neutralization test is designed to test for antibodies that provide protection from the effects of pathogenic agents or toxins. For this test, a known toxic antigen or organism is used with the test serum from an animal being tested, which may or may not have protective antibodies. Two groups of mice are administered just enough toxic antigen or organism to kill the mice. Antiserum from an animal being tested is injected into one of the groups of mice. If the mice from the group given the antiserum survive but the mice not given serum die or become ill, this is evidence that the antiserum contained protective antibody. This protocol has been used to test for antibodies against a variety of toxic agents, especially viruses (Parker and Thompson 1942, Lancaster 1966, Plowright 1981, Seymour and Yuill 1981).
Variations of the neutralization test also can be used to test for the antigens of a toxin or toxic agent using known antibodies (antisera). One the first of such tests, the mouse protection test (Quortrup and Sudheimer 1943), is used for the diagnosis of botulism (intoxication from the bacterium Clostridium botulinum) (Rocke and Bollinger 2007). In this test, blood serum from a sick or dead bird is injected into two groups of mice, one of which previously was protected with type-specific antibodies (e.g., botulism type C antitoxin). Botulism toxin is considered present in the serum if the protected mouse survives and the unprotected mouse develops paralysis or other symptoms characteristic of botulism (Rocke and Bollinger 2007). This test increasingly is being replaced by ELISA tests, which are both cheaper and avoid the use of live animals (Thomas 1991, Rocke and Bollinger 2007).
Complement Fixation Test
The complement fixation (CF) test is based on the recognition that complement is an integral component of antigen-antibody reactions. Complement is loosely bound by antibody, and firmly bound by antigen-antibody complexes. The CF test is based on having two antigen-antibody systems compete for a limited amount of complement. One, the test system, comprises the infective agent or toxic antigen of interest along with sera of animals being tested for the presence of antibody to that antigen. The second system, called the indicator system, comprises sheep erythrocytes and hemolysin; hemolysin is an antibody made against sheep erythrocytes in rabbits. Hemolysin combines with sheep erythrocytes to form “sensitized red cells,” but causes no observable effect. However, in the presence of complement, these sensitized red cells lyse. In the CF test, there is only enough complement available to allow one of these antigen-antibody reactions to occur.
In conducting the test, the test system of antigen and the unknown serum first are combined with complement.
Then the indicator system comprising the sheep erythrocyte-hemolysin mixture (sensitized red cells) is added. If the unknown serum being tested has a specific antibody to the known antigen, the complement is tied up in the test system and no complement is available for the indicator system of sheep erythrocytes and hemolysin; thus no hemolysis of sensitized red cells occurs when that system is added. However, if the specific antibody is not present in the unknown serum, complement is not tied up by the test system and thus becomes available to cause the lysis of sheep erythrocytes in the indicator system. Thus hemolysis is the endpoint of a negative test for the antibody being tested, and absence of hemolysis is the endpoint for a positive test. The CF test has been adapted to many different antigen-antibody tests because the sensitivity is considered very high; thus it is a valuable test despite its complexity. Examples of its use include some bacteria (Rice 1961b, Cavanaugh et al. 1965) and many viruses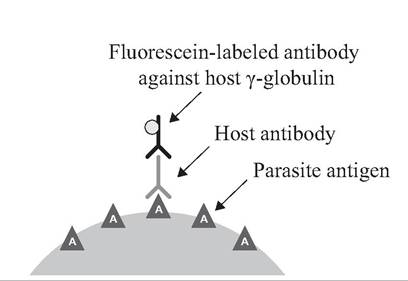
FIGURE APP. 2.1 Indirect fluorescent antibody (IFA) test.
(Rice 1960a, 1960b, 1960c, 1960d, 1960e, 1960f, 1961a, 1961c; Seymour and Yuill 1981).
Fluorescent Antibody Tests
The indirect fluorescent antibody (IFA) test (Food and Agricultural Organization of the United Nations. 1998), also called the indirect immunofluorescence test, is a test for antibodies against a specific antigen using an indicator dye that fluoresces under ultraviolet light (Fig. App. 2.1). There are four chief components for the test. One is a purified preparation of the known antigen (e.g., rabies virus). Another is the antisera of the animals being tested (e.g., foxes); these antisera may (or may not) have the antibodies being sought. One also needs antibodies from another animal (e.g., laboratory rabbit) made against the host (e.g., fox) γ-globulin. Finally, there is a fluorescent dye such as fluorescein isothiocyanate, which serves as the indicator of a positive test.
For the IFA test, antibodies are made against the γ-globulin of the host species (e.g., fox) whose sera are being tested for antibodies (e.g., anti-rabies); one can produce antibodies against fox γ-globulin in laboratory rabbits. The fluorescent dye then is chemically conjugated to the rabbit anti-fox γ-globulin and stored. A separate slide with a fixed smear of the known antigen (e.g., rabies virus) is flooded with each fox serum being tested. If anti-rabies antibodies are present in a serum, they combine with the rabies antigen on the slides in a stable antigen-antibody reaction. There is no visible change, however. All excess unattached antibodies are rinsed off. Next, anti-fox γ-globulin antibodies with the conjugated fluorescent dye are added to each slide. For serum samples with fox antibodies adhering to the rabies antigen on the slide, these fox antibodies (γ-globulin) now serve as antigens for rabbit antibody against fox γ-globulin; the anti-fox γ-globulin antibody attaches, bringing along the conjugated fluorescent dye. For fox serum samples with no antibodies against rabies, the extraneous antibodies are rinsed off and there is no fox γ-globulin available to serve as an antigen for anti-fox γ-globulin antibodies; hence, there is no visible fluorescence. The presence of the fluorescent dye under an ultraviolet microscope is the sign of a positive test for antibodies against rabies in a fox sample. Although complex, this is a very sensitive method for antibody detection, and generally is reported simply as positive or negative for the antibody presence. This test can be used for many antigens.
The direct fluorescent antibody (DFA) test is used for the direct detection of antigens, such as rabies virus in the brain smear of a suspect animal, using known fluorescent-labeled rabies-specific antibodies (Fig. App. 2.2). The use of monoclonal antibodies can add specificity to the test.
Enzyme-linked Immunosorbent Assay (ELISA)
The enzyme-linked immunosorbent test (ELISA) is an immunoassay using an enzyme reaction as an endpoint (Janeway and Travers 1997, Lapointe et al.
2000, Tizard 2004) (Fig. App. 2.3). Enzyme immunoassays typically use visible enzyme-mediated reactions; older tests used color-changed products of enzymesubstrate interaction, whereas newer assays employ fluorogenic substrates that have a much higher sensitivity (Tizard 2004). The resulting chromogenic, fluorogenic, or electrochemical changes can be measured with a spectrophotometer, spectrofluorometer, or other optical/electrochemical device to provide a quantitative estimate of antibody in a serum sample (Coleman et al. 1989).Four components are needed for the indirect ELISA assay, a measure of antibody in a sample. One component is a known antigen preparation (e.g., deer adenovirus) (Lapointe et al. 2000). Another is a serum sample of the animal being tested (e.g., deer), which may or may not have specific antibodies against the adenovirus antigen. A third component is a supply of antibodies from another animal (e.g., lab rabbit) made specifically against deer γ-globulin. Finally, one needs a substrate-enzyme system that, when combined, will produce a chromogenic, fluorogenic, or electrochemical signal (Tizard 2004).
To conduct the indirect assay, the enzyme is attached to the purified antibody (e.g., rabbit) against the γ-globulin of the host (e.g., deer) being tested. A quantified sample of known (e.g., adenovirus) antigen is applied to the well of a polystyrene microtiter plate. One host (deer) serum sample to be tested is added to each of the wells of a microtiter plate; any antibodies against the pathogen being tested (adenovirus) combine specifically with the antigen attached to the surface of the well. The enzyme-labeled rabbit (antideer) antibody is added to the wells, where they bind to any deer γ-globulin; nonspecific binding is inhibited. Unbound labeled antibody is removed from wells by
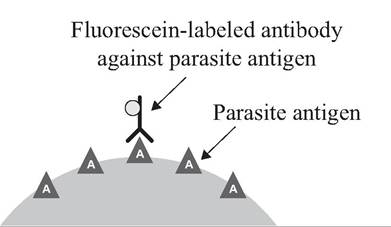
FIGURE APP. 2.2 Direct fluorescent antibody (DFA) test.
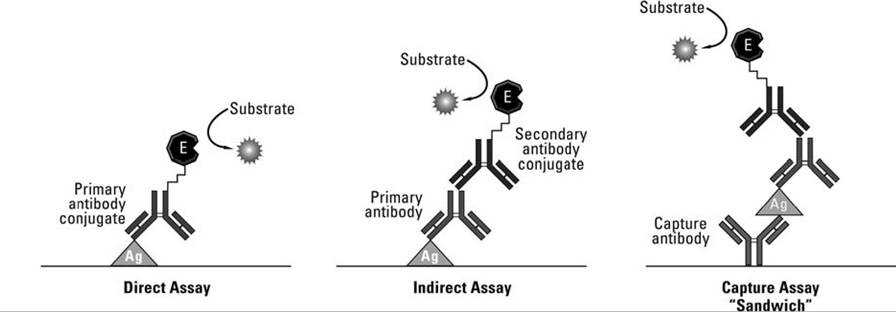
FIGURE APP. 2.3 Three common ELISA techniques. Ag: known antigen; Primary antibody: specific antibody (e.g. against adenovirus) being tested from a host species (e.g., deer); Secondary antibody: known antibody against γ-globulin of host (e.g., deer) being tested for antibody; E: substrate specific enzyme producing a detectable chromogenic response in the presence of the added substrate (Courtesy of Thermo Fisher Scientific).
thorough rinsing with a mild detergent. One then adds a colorless substrate of the enzyme to elicit a color, fluorescent, or electrochemical reaction. The intensity of the visible response when the substrate is cleaved by the enzyme attached to the detecting antibody is directly correlated to the amount of specific antibody to the adenovirus in the unknown deer serum.
The ELISA technique also can be adapted for detection and quantification of antigen in a test sample (Tizard 2004). In the direct assay (Fig. App. 2.3), the known specific antibody for the antigen being tested is labeled with the enzyme directly. The unknown antigen being tested is coated on a microtiter plate. The labeled antibody for that antigen is flooded over the antigen, excess antibody is washed off, and the resulting color or fluorescence can be quantified. The test is relatively quick and avoids potential cross-reactivity, but does require the labeling of the specific antibody for each host tested, which can be time consuming and expensive.
A common test for antigen is the capture (sandwich) assay (Fig. App. 2.3). Here, unlabeled specific antibodies to the antigen sought first are bound to the well of a microtiter plate. When an unknown antigen sample is added, it will attach to the specific antibodies lining the well if it is the antigen sought; other antigens are washed away. Unlabeled specific antibodies for the antigen sought are again added, followed by an enzyme-labeled antibody to the Fc section of this middle layer of antibody (the Fc portion is the base or bottom of the Y-shaped antibody). Adding the test enzyme substrate gives a visible indicator ofthe antigen's presence; further, the amount of antigen is correlated to the intensity of the visible change. An advantage of the “sandwich ELISA” is that one can use relatively crude and impure antigenic samples and still readily detect the presence of the antigen sought.
One also can detect antigen by the competitive ELISA assay; unlabeled specific antibody to the antigen sought first is coated onto the wall of the microtiter well. A sample of antigen to be tested is added to each well. If the specific antigen sought is present, it will bind with the known antibody coating the well. After that reaction is completed, an enzyme- labeled (known) antigen is added to the well, which then attaches to any remaining antibody-binding sites (Tizard 2004). The substrate for the enzyme is added to elicit the visible response. The more antigen that was present in the original test sample, the fewer antibody-binding sites will be available for the enzyme-labeled known antigens in the coated well, and the weaker the chromogenic or fluorogenic signal when the enzyme acts on the substrate. Thus, a weak or negative test is evidence for presence of specific antigen sought in the test sample that competed with the known, labeled antigen subsequently added. In contrast, a strong positive is evidence that there was little of the antigen sought in the test sample to compete with the labeled known antigen.
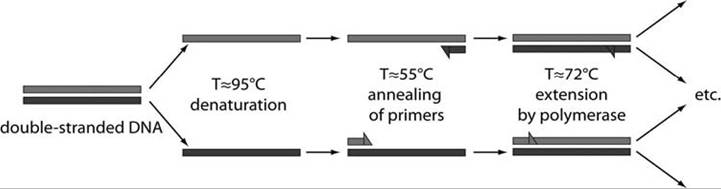
FIGURE APP. 2.4 Outline ofthe classical polymerase chain reaction (PCR) schematic, including denaturation of DNA, annealing of primers, and extension of the sample DNA by polymerase (Courtesy of Juan Santiago, Stanford University, California).
Gel Electrophoresis
Gel electrophoresis is a technique used for the separation of protein molecules or nucleic acids in a gel matrix, using an electric current applied to the gel matrix (Berg et al. 2002). Electrophoresis refers to the electromotive force used to move the molecules through the gel matrix.
In gel electrophoresis, mixtures of molecules in an agarose gel are added into wells on a polyacrylamide plate or similar polymer. Electric current is passed through the gel. The molecules migrate based on their charges and size. Positive ions are pulled to the negative pole; smaller molecules move faster than large ones.
The isolated clusters of molecules then are exposed to a stain or an imaging molecule (e.g., Coomasie blue dye, ethidium bromide, fluorescent markers, radiolabels) and are photographed or viewed under ultraviolet light. Radiolabels exposed to film are called autoradiograms.
Western Blot Test
The western blot test, also known as the immunoblot test, is used to detect a specific protein in a given sample of tissue homogenate or extract (Tizard 2004). It uses gel electrophoresis to separate native or denatured proteins, based on the length of the polypeptide or the 3-dimensional structure of the protein. The original mixtures of proteins are loaded into wells on an agarose plate. An electric current is passed through the gel to separate them. After a standard time, proteins with similar molecular weight and charge will group close together on the gel. These proteins then are “blotted” onto a membrane such as nitrocellulose, retaining their electrophoretic positions. Following this, they are combined with specific antibodies against target proteins on the blot. The bound antibodies are then identified using an enzyme immunoassay such as ELISA. The name western blot is a play on the name Southern blot, a technique for DNA detection (Burnette 1981) developed earlier by Edwin Southern; detection of RNA is termed northern blotting.
Immunohistochemistry
Immunohistochemistry (IHC) refers to the process of localizing proteins in cells of a tissue section using antibodies that bind specifically to those protein antigens in biological tissues (Ramos-Vara 2005). Unlabeled specific antibody is used to combine with a target antigen in complex biological samples such as histologic sections of tissues. A second antibody, usually with a marker or enzyme bound to it, is used as a detector. There generally are two strategies used for immunohistochemical detection of antigens in tissue: the direct method and the indirect method (Tizard 2004).
The direct method involves a one-step staining methods with a labeled antibody (e.g., fluorescein isothiocyanate conjugate) reacting directly with the antigen in tissue sections. With only one antibody involved, this procedure is simple and rapid but has less sensitivity in detecting the antigen sought.
The indirect method involves an unlabeled primary antibody (first layer), which reacts with the target tissue antigen and a labeled secondary antibody is made against the IgG of the animal species from which the primary antibody is derived. The secondary layer antibody is labeled with an enzyme or fluorescent dye for detection.
Polymerase Chain Reaction
The polymerase chain reaction (PCR) is a molecular, rather than serologic, process used to exponentially amplify low levels of specific DNA sequences in a serum or tissue sample to a threshold level where that DNA sequence can be established (Janeway and Travers 1997) (Fig. App. 2.4). One starts with the serum or other sample containing the genetic material to be amplified. That DNA sequence can be a single gene, just a part of a gene, or a non-coding sequence. For each specific DNA sequence being amplified, the PCR requires two known primers (20-30 base fragments of DNA) complementary to the ends of that specific sequence. One then adds short, known portions of these single DNA strand primers that are complementary to both the 5' and 3' ends of specific DNA sequence being amplified. A heat-stable polymerase such as Taq polymerase, named after the heat-stable bacterium Thermus aquaticus, is used to synthesize copies of the DNA region to be amplified. Deoxynucleotide triphosphates are added, from which the DNA polymerase builds the new DNA.
Using a thermal cycler to heat and cool the three key steps, the double DNA strands first are heated to about 95°C to disrupt the hydrogen bonds and produce single-stranded DNA molecules. The temperature then is lowered to 55°C to let the primers anneal to the single-stranded DNA. The Taq polymerase binds to the primer-template hybrid and begins DNA formation. With the temperature at 72°C, the polymerase synthesizes a new DNA strand complementary to the single-stranded DNA.
Each denaturation-annealing cycle doubles the amount of the specific DNA sequence being amplified and the cycles are continued 30 or more times; thus the process produces millions of copies of a single DNA segment in a matter of hours. These resulting DNA fragments are hybridized to a radio- actively labeled DNA segment complementary to a short sequence of the amplified DNA. Following electrophoresis, the radio-labeled product of specific size is detected by autoradiography. This test commonly is used to identify current presence of the DNA for an infectious agent; presence of that DNA is strong evidence of infection by that agent at the time of the sample. It is also possible to apply the PCR test to fecal samples, from which one can isolate mitochondrial DNA, microsatellite DNA, and single-copy nuclear DNA, and thus determine considerable information about the species and even individual characteristics of the host in question (Wasser et al. 2002). There are numerous variations of the PCR test.
Quantitative Real-time Polymerase Chain Reaction
One variation, the quantitative real-time polymerase chain reaction (QRT-PCR), or kinetic polymerase chain reaction, is used to amplify and simultaneously quantify a targeted DNA molecule. It enables detection of a specific sequence in a DNA sample, either as an absolute number of copies or as a relative amount (Logan et al. 2009). Quantitative real-time PCR quantifies DNA in a sample by measuring fluorescent markers after each cycle. By having a fixed cap on how much DNA is being produced, and knowing how many cycles are required to produce that cap, one can extrapolate back to estimate how much DNA must have been present in the original sample. Advantages include good analytical sensitivity, fast results, and broad applicability.
DNA Sequencing
Use of DNA sequencing encompasses biochemical methods for determining the order of the nucleotide bases in a DNA oligonucleotide. A wide variety of techniques have been employed since the development of recombinant DNA technology in 1972. The various techniques used provide the sequence of nucleic acids along a piece of DNA; they can be used to identify products from PCR reactions and sometimes entire genomes (Kieleczawa 2005).
Restriction Fragment Length Polymorphism
Restriction fragment length polymorphism (RFLP) is a technique used to identify differences in the nucleic acid sequence that occur at a site where a restriction enzyme cuts (Botstein et al. 1980). It is an important technique in genetic fingerprinting. Purified DNA is cut into restriction fragments using suitable endonucleases, which only cut the DNA molecule at specific DNA sequences, termed recognition sequence or restriction sites, that are recognized by the enzymes. These sequences are specific to each enzyme and may be 4, 6, 8, 10, or 12 base pairs in length. Restriction fragments are separated by length and molecular weight with agarose gel electrophoresis. The distance between locations cut by the restriction enzymes varies among individuals, resulting in varying fragment lengths; thus, one can distinguish between individuals and also assess genetic relatedness among individuals. In wildlife diseases, the RFLPs can be used to associate genetic characteristics of pathogens with patterns of virulence or host associations.
LITERATURE CITED
Bell, J. F. 1981. Q fever. Pp. 388-397 in J. W. Davis,
L. H. Karstad, and D. O. Trainer (editors), Infectious diseases of wild mammals. 2nd ed. Iowa State University Press, IA.
Berg, J. M., J. L. Tymoczko, and L. Stryer. 2002.
Molecular cell biology. W. H. Freeman, New York.
Botstein, D., R. L. White, M. Skolnick, and R. W. Davis. 1980. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American Journal of Human Genetics 32:314-331.
Burnette, W. N. 1981. “Western blotting”: Electrophoretic transfer of proteins from sodium dodecyl sulfate—polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Analytical Biochemistry 112:195-203.
Cavanaugh, D. C., B. D. Thorpe, J. B. Bushman, P. S. Nicholes, and J. J. H. Rust. 1965. Detection
of an enzootic plague focus by serological methods. Bulletin of the World Health Organization 32:197-203.
Chernesky, M. A. 1996. Traditional serological tests. Pp. 107-123 in B. W. J. Mahy and H. O. Kangro (editors), Virology methods manual. Academic Press, London, UK.
Coleman, R. M., M. F. Lombard, R. E. Sicard, and
N. J. Rencricca. 1989. Fundamental immunology. Wm. C. Brown, Dubuque, IA.
Damon, S. R., and M. B. Johnson. 1944. A rapid agglutination test for the diagnosis of tularemia. Journal of Laboratory and Clinical Medicine 29:976-977.
Food and Agricultural Organization of the United Nations. 1998. A field guide for the diagnosis, treatment and prevention of African animal try- panosomosis. Food and Agricultural Organization of the United Nations, Rome, Italy.. Figure 14, Immunofluorescent antibody test, available at.
Hedger, R. S. 1981. Foot-and-mouth disease. Pp. 87-96 in J. W. Davis, L. H. Karstad, and
D. O. Trainer (editors), Infectious diseases of wild mammals. 2nd ed. Iowa State University Press, Ames, IA.
Hermann, J. E. 1995. Immunoassays for the diagnosis of infectious diseases. Pp. 110-122 in
P. R. Murray, E. J. Baron, M. A. Pfaller,
F. C. Tenover, and R. H. Yolken (editors), Manual of clinical microbiology. 6th ed. ASM Press, Washington, DC.
Hierholzer, J. C., M. T. Suggs, and E. C. Hall. 1969. Standardized viral hemagglutination and hemagglutination-inhibition tests. Applied Microbiology 18:824-833.
Janeway, C. A., and P. Travers. 1997. Immunobiology: The immune system in health and disease. Current Biology, London, UK.
Kaskara, Y. 1997. Agglutination immunoassays.
Pp. 7-12 in N. R. Rose, E. C. deMacario,
J. D. Folds, J. C. Lane, and R. M. Nakamura (editors), Manual of clinical laboratory immunology. 5th ed. ASM Press, Washington, DC.
Kieleczawa, J. (editor). 2005. DNA sequencing: Optimizing the process and analysis. Jones & Bartlett Publishers, Sudbury, MA.
Lancaster, J. E. 1966. Newcastle disease: A review of the literature published between 1926 and 1964. Monograph No. 3. Canada Department of Agriculture, Ottawa, Canada.
Lapointe, J.-M., L. W. Woods, H. D. Lehmkuhl,
M. K. Keel, P. V. Rossitto, P. K. Swift, and
N. J. MacLachlan. 2000. Serologic detection of adenoviral hemorrhagic disease in black-tailed deer in California. Journal of Wildlife Diseases 36:374-377.
Ledingham, J. C. G. 1937. Studies on the serological interrelationships of the rabbit viruses, myxomatosis (Sanarelli, 1898), and fibroma (Shope, 1932). British Journal of Experimental Pathology 18:436-449.
Logan, J., K. Edwards, and N. Saunders (editors). 2009. Real-time PCR: Current technology and applications. Caister Academic Press, Norwich, UK.
Mahony, J. B., and M. A. Chernesky. 1999. Immunoassays for the diagnosis of infectious diseases. Pp. 202-214 in P. R. Murray, E. J. Baron, M. A. Pfaller, F. C. Tenover, and R. H. Yolken (editors), Manual of clinical microbiology. 7th ed. ASM Press, Washington, DC.
Ouchterlony, O. 1958. Diffusion-in-gel methods for immunological analysis. Progress in
Allergy 5:1-78.
Parker, R. F., and R. L. Thompson. 1942. Neutralization of virus of myxoma by specific immune serum. Journal of Immunology 40:147.
Plowright W. 1981. Herpesvirus of wild ungulates, including malignant catarrhal fever virus. Pp. 126-146 in J. W. Davis, L. H. Karstad, and D. O. Trainer (editors), Infectious diseases of wild mammals. 2nd ed. Iowa State University Press, Ames, IA.
Quortrup, E. R., and R. L. Sudheimer. 1943. Detection of botulinus toxin in the blood stream of wild ducks. Journal of the American Veterinary Medical Association 102:264-266.
Ramos-Vara, J. A. 2005. Technical aspects of immunohistochemistry. Veterinary Pathology 42:405-426.
Rice, C. E. 1960a. Introduction to the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part I: Introduction. Canadian Journal of Comparative Medicine and Veterinary Science 24:126-130.
Rice, C. E. 1960b. The use of the complement-fixation test in the study and diagnosis of viral diseases in man and animals: A review. Part II: The pox viruses. Canadian Journal of Comparative Medicine and Veterinary Science 24:254-257.
Rice, C. E. 1960c. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part III: Vesicular diseases. Canadian Journal of Comparative Medicine and Veterinary Science 24:204-208.
Rice, C. E. 1960d. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part III: Vesicular viruses (cont'd). Canadian Journal of Comparative Medicine and Veterinary Science 24:238-241.
Rice, C. E. 1960e. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part IV: Some neurotropic viruses. Canadian Journal of Comparative Medicine and Veterinary Science 24:301-305.
Rice, C. E. 1960f. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part V: The arboviruses. Canadian Journal of Comparative Medicine and Veterinary Science 24:352-358.
Rice, C. E. 1961a. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part VI: The enteroviruses. Canadian Journal of Comparative Medicine and Veterinary Science 25:35-41.
Rice, C. E. 1961b. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part VII: The psittacosis-lymphogranuloma venereum group. Canadian Journal of Comparative Medicine and Veterinary Science 255:74-79.
Rice, C. E. 1961c. The use of the complementfixation test in the study and diagnosis of viral diseases in man and animals: A review. Part VIII: The myxoviruses. Canadian Journal of Comparative Medicine and Veterinary Science 25:151-156.
Rocke, T. E., and T. K. Bollinger. 2007. Avian botulism. Pp. 377-416 in N. J. Thomas, D. B. Hunter, and C. T. Atkinson (editors), Infectious diseases of wild birds. Blackwell Publishing, Ames, IA.
Seymour, C., and T. M. Yuill. 1981. Arboviruses. Pp. 54-86 in J. W. Davis, L. H. Karstad, and D. O. Trainer (editors), Infectious diseases of wild mammals. 2nd ed. Iowa State University Press, Ames, IA.
Thal, E. 1956. Untersuchungen uber Pasteurella pseudotuberculosis unter besonderen Berucksich- tigung ihres immunologischen Verhaltens. Berlingska Boktrycheriet, Lund, Sweden.
Thomas, R. J. 1991. Detection of Clostridium botulinum type C and D toxin by ELISA. Australian Veterinary Journal 68:111-113.
Tizard, I. R. 2004. Veterinary immunology: An introduction. Saunders/Elsevier, Philadelphia, PA.
Vaananen, P., V. M. Haiva, P. Koskela, and
O. Meurman. 1985. Comparison of a simple latex agglutination test with hemolysis-in-gel, hemagglutination inhibition, and radioimmunoassay for detection of rubella virus antibodies. Journal of Clinical Microbiology 21:793-795.
Wasser, S. K., K. E. Hunt, and C. M. Clarke. 2002. Assessing stress and population genetics through noninvasive means. Pp. 130-144 in A. A. Aguirre, R. S. Ostfeld, G. M. Tabor, C. House, and M. C. Pearl (editors), Conservation medicine: Ecological health in practice. Oxford University Press, New York.
Wetzler, T. F., H. E. Eitzen, J. A. Currie, and J. J. D. Marshall. 1968. Lipopolysaccharide-like antigens from Pasteurella pseudotuberculosis shared by various genera of Enterobacteriaceae as demonstrated by hemagglutination tests. Symposia Series in Immunobiological Standardization 9:155.
Witter, J. F. 1981. Brucellosis. Pp. 280-287 in J. W. Davis, L. H. Karstad, and D. O. Trainer (editors), Infectious diseases of wild mammals. 2nd ed. Iowa State University Press, Ames, IA.
Wright, G. G., and R. J. Feinberg. 1952. Hemagglutination by tularemia antisera: Further observations of agglutination of polysaccharide-treated erythrocytes and its inhibition by polysaccharide. Journal of Immunology 68:65-71.
More on the topic DIAGNOSTIC TECHNIQUES:
- PCR Techniques
- Evaluation of Diagnostic Tests Under Local Conditions
- Chapter 48 Modern Risk Management Techniques in Banking Sector
- Commercial Diagnostic Kits
- A POTPOURRI OF LIBERAL REVENUE RECOGNITION TECHNIQUES
- AEROSOL DEVICES AND TECHNIQUES
- Diagnostic Tests
- Sample-Specific Techniques and Challenges
- Sectoral Institutions and Techniques of Control
- DIAGNOSTIC APPROACH IN CHD
- Application and Interpretation of Findings From PCR-Based Diagnostic Tests
- Quality Control of PCR-Based Diagnostic Assays
- DIAGNOSTIC APPROACH IN NEONATAL JAUNDICE
- Performance of various diagnostic and therapeutic procedures on children
- Europeans did not deploy everywhere the full battery of institutions and techniques noted in chapter 12.
- DIAGNOSTIC ASSESSMENT