NEURAL TUBE DEFECTS
Neural tube defects (NTDs) is a collective term for broad spectrum of developmental defects, due to defective
TABLE 18.24: Developmental CNS malformations
• Defective formation of neural tube (Neural tube defects) Spina bifida occulta
MeningoceleZMyelomeningocele
Encephalocele
Anencephaly
• Defective segmentation of neural tube
Holoprosencephaly
Porencephaly
Agenesis of corpus callosum
• Disorders of neural migration or sulcation
Lissencephaly
Schizencephaly
• Disorders of neurological growth
Microcephaly
Hydrocephalus
Craniosynostosis
fusion of neural tube, ranging from asymptomatic spina bifida occulta to most severe anencephaly and encephalocele.
Overall incidence of NTDs in India is estimated to be ~ 1.5/1000 live births, more common in north India and Punjabi population (4-8/1000).
Etiology: While some NTDs are part of chromosomal disorders, e.g. Trisomy 13 (Patau syndrome), isolated NTDs are multifactorial in origin. Dietary folates and folic acid also have an important role in causation of NTDs.
Important maternal risk factors for NTDs include: (a) folic acid deficiency, (b) severe malnutrition, (c) alcohol abuse, (d) radiation exposure, (e) diabetes mellitus and (f) anticonvulsant-exposure, e.g. valproate or carbamazepine.
Clinical spectrum varies according to the type of defect, as follows:
a. Meningomyelocele is the commonest identified neural tube defect (NTDs), characterized by protusion of meningies and neural tissue remnants through a midline vertebral column defect (dysraphism), with or without secondary neurological damage. Clinical features include:
± Posterior midline cystic (translucent) mass, usually in lumbosacral region, covered with thin layer of partially epithelialized tissue (Fig. 18.4).
± Secondary neurological damage depending on location of the lesion, e.g.
paraplegia with/without sensory damage and bladder/bowel involvement± Associated abnormalities, e.g. (a) Arnold-Chiari malformation with hydrocephalus, (b) genitourinary tract abnormalities, and (c) skeletal defects, e.g. club feet and congenital dislocation of hip.
b. Meningocele is the milder variant, in which herniated sac contains only meninges and no neural
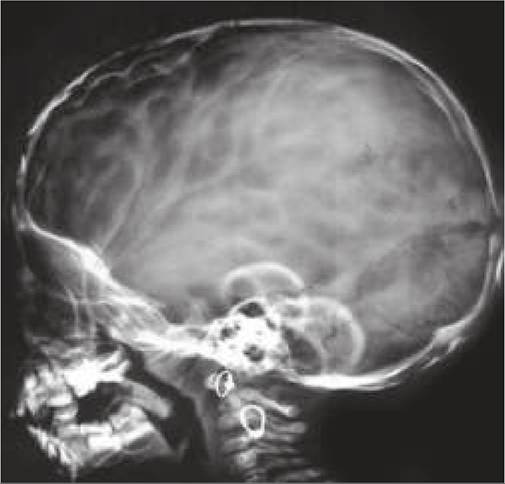
Fig. 18.4: Meningomyelocele.
tissue. However, associated abnormalities, e.g. syringomyelia, diastematomyelia, tethered cord syndrome and hydrocephalus are not uncommon and should be excluded by MRI, before surgery.
c. Encephalocele is the cranial variant of the meningocele, characterized by herniation of a meningial sac with/ without brain matter, from occipital or frontal region. Associated malformations include hydrocephalus, Arnold-Chiari malformation, Dandy-Walker cysts, etc.
d. Anencephaly is the most severe NTD, characterized by a large defect in calvarium, meninges and scalp with rudimentary brain, due to failure of closure of the rostral neuropore. The primitive brain usually contains only residual brain-stem, without cerebrum, cerebellum and other structures. Polyhydramnios is common. Child usually succumbs in few days.
e. Spina bifida occulta is the commonest, though frequently missed NTD, characterized by midline defect in vertebral bodies covered with skin without protrusion of spinal contents, usually at L5-S1. Presence of a tuft of hair, dermal sinus, discoloration or lipoma over spine indicates possibility of underlying spina bifida occulta. Presence of dermal sinus with spina bifida occulta may lead to recurrent meningitis.
Diagnosis of NTDs is clinically obvious, except in spina bifida occulta, but further investigations are essential to exclude other problems, e.g. (a) Neuroimaging of entire spine and brain to exclude hydrocephalus, and (b) Renal USG to exclude congenital anomalies and bladder dysfunction, which are common in these cases.
Neuroimaging (MRI) is indicated even in spina bifida occulta to exclude cord defects, e.g. syringomyelia, diastematomyelia and tethered cord syndrome.
Prenatal diagnosis of NTDs is possible by 16-18 weeks, on antenatal USG or presence of following biochemical markers:
• Elevated #945;-fetoprotein levels in maternal serum with 60-70% sensitivity;
• Elevated #945;-fetoprotein or acetylcholinesterase levels in amniotic fluid, with ~100% sensitivity.
Management of NTDs depends on the severity of defect, presence of secondary neurological deficits and other congenital anomalies. Immediately at birth, open defects should be covered with sterile wet gauze to prevent drying, cracking and infection of covering tissue, till surgery is arranged.
CSF examination and antibiotics are indicated in cases with ruptured membranes with CSF leak or infection, in whom surgery should be deferred till the CSF is sterile.
Early surgical repair of defect within 24-48 hours of birth along with CSF shunt (in cases with associated hydrocephalus) is indicated in selected cases of myelomeningocele. Prenatal in utero closure is possible at few centres, with better functional results.
However, surgery is usually futile in cases with-a) severe paraplegia, (b) gross hydrocephalus or encephalo- cele, (c) gross kypho-/scoliosis, (d) serious renal or other congenital anomalies (Lorber's criteria).
No intervention is required for spina bifida occulta, unless associated with other defects. malformations, e.g. cyclopia (single mid-line eye), cebacephaly and premaxillary agenesis is common. Most cases die in infancy (Fig. 18.5).
Porencephaly is the presence of cysts/cavities in cerebral cortex, which may be congenital or acquired after cerebral infarcts or hemorrhage. Congenital cysts are often CSF- filled, due to communication with subarachnoid or ventricular spaces and frequently symptomatic with focal seizures and hemiparesis (Fig. 18.5).
Hydranencephaly represents an extreme form of porencephaly with near absence of cerebral hemispheres, probably related to bilateral occlusion of internal carotid arteries in early fetal life.
Head size is normal at birth but enlarges rapidly with positive transillumination.Clinically, these cases present with large head, severe developmental retardation, seizures and spastic quadriparesis. CSF shunt may prevent progression of head enlargement (Fig. 18.5).
Outcome depends on presence of neurological deficit and hydrocephalus at the time of surgery. With early surgery in cases without neurological deficit, prognosis is good, though ~10-15% cases may die due to meningitis, late hydrocephalus or other malformations.
Survivors may be normal or left with—(a) paraplegia, (b) bladder/bowel incontinence, (c) seizure disorders and (d) learning disabilities or intellectual dusability.
Prevention of NTDs is a major advance in prenatal therapeutics, which involves:
• Genetic counselling regarding risk of recurrence in future pregnancy, which is ~5% (if one parent/sibling is affected) and ~10 % (if two siblings are affected).
• Folic acid supplements: All prospective mothers with history of previous NTD birth, should receive 5 mg/day of folic acid, from one months before conception till the end of first trimester, which reduces the risk of recurrence by gt;50%.
Primary prevention to all prospective mothers irrespective of high-risk history with lower dose of folic acid supplementation (0.4 mg/day) during this period is also recommended at present, which can reduce the risk of NTD by gt;70%.
• Prenatal diagnosis with USG/biochemical markers, discussed earlier, with appropriate interventions.
Some other important developmental CNS malformations are as follows:
Neural tube segmentation defects are relatively uncommon and include:
Holoprosencephaly is characterized by incomplete separation of two cerebral hemispheres with single ventricle, absence of falx cerebri and fused basal ganglion, due to defective cleavage of prosencephalon. Associated
Fig.
18.5: Important development malformations of brain: (A) Holoprosencephaly; (B) Porencephaly; (C) Hydranen- cephaly; (D) Agenesis of corpus callosum; (E) Lissencephaly; (F) SchizencephalyAgenesis of corpus callosum is either isolated or associated with other neurological or chromosomal disorders, e.g. Aicardi syndrome. These cases may be entirely asymptomatic with accidental diagnosis on CT/MRI scan or may present with diplegia, mental retardation and seizures (Fig. 18.5).
Neuronal migration defects are easily missed on CT scan and being increasing recognized as a cause of unexplained mental retardation or refractory seizures, after advent of MRI.
Lissencephaly (agyria) is characterized by the absence of cerebral convolutions and poorly formed sylvian fissure, due to faulty migration of neuroblasts in early embryonic life. These cases present with microcephaly, severe mental retardation, seizures and failure to thrive in infancy (Fig. 18.5).
Schizencephaly is characterized by presence of unilateral or bilateral clefts in cerebral hemispheres, presenting with seizures, severe mental retardation and spastic quadriparesis in bilateral lesions (Fig. 18.5).
Microcephaly denotes head circumference lt;3rd percentile for corresponding age and sex.
Etiologically, it may be primary, associated with genetic disorders; or secondary due to intrauterine or postnatal insult to growing brain (Table 18.25).
Clinically, microcephaly is almost always associated with mental retardation with/without seizures and features of chromosomal defects/intrauterine infections abnormal facial appearance with slanting forehead and prominent nose and ears is more common in primary microcephaly (d/d secondary microcephaly).
Diagnosis rests on head circumference, though etiological diagnosis requires neuroimaging, karyotyping/ genetic testing and serological studies for intrauterine infections.
D/D of microcephaly includes craniosynostosis-a cranial vault defect with premature fusion of sutures, leading to abnormal head shapes, restricted brain growth and neuroocular abnormalities (see Table 23.2).
Management of these cases is mainly directed towards the primary cause, though outcome is generally unsatisfactory (Fig. 18.6).
TABLE 18.25: Common causes of microcephaly
Primary microcephaly
• Genetic: Familial (AR) or autosomal dominant
• Chromosomal: Trisomies, cri-du-chat syndrome
Secondary microcephaly
• Intrauterine infections: Rubella, CMV
• Intrauterine radiation/drugs: Alcohol, hydantoin
• Birth asphyxia: Hypoxic-ischemic encephalopathy
• Perinatal CNS infections: Meningitis
• Severe malnutrition (?)
• Metabolic disorders, e.g. Phenylketonuria
Fig. 18.6: Microcephaly.
18.10.2
More on the topic NEURAL TUBE DEFECTS:
- NEURAL TUBE DEFECTS
- Extensive neuropathologic studies have demonstrated that neural tube defects are associated with a high incidence of gross and microscopic malformations of the forebrain and hindbrain (34).
- Cerebellum and Hindbrain
- TECHNICAL FACTORS OF NEEDLE ELECTROMYOGRAPHY
- Structural abnormalities
- Diabetes mellitus
- Universal screening and case finding
- PRECONCEPTION AND interconception care ^292
- Endocrinology
- Lifestyle Considerations for Multiple Pregnancy