Secret Operations: Intestinal Epithelial Cells, Macrophages and MAP
Sun Tzu writes that ‘Secret operations are essential in war; upon them the army relies to make its every move’ (Tzu, 1971, p. 149). The ability of MAP to infiltrate and enter host cells without overtly alarming the immune system may explain why it is so well adapted to its ruminant host (Fig.
9.1). As previously mentioned, MAP is spread by the faecal-oral route and gains entry to intestinal walls through the small intestinal mucosa via M cells or villous epithelial cells overlying Peyer’s patches in gut-associated lymphoid tissue (SigurSardottir et al., 1999, 2004; Whittington and Sergeant, 2001; Whittington et al., 2004; Crossley et al., 2005; Grewal et al., 2006; Tiwari et al., 2006). M cells represent a primary target for MAP infection, which may be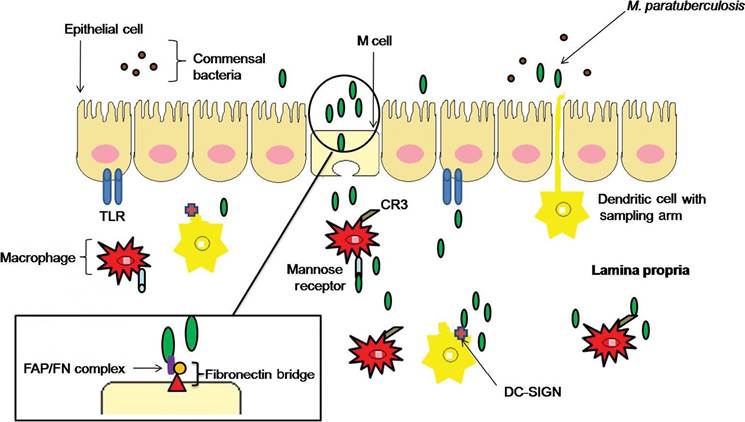
Fig. 9.1. Intestinal cell wall and macrophage invasion tactics used by Mycobacterium avium subsp. paratuberculosis (MAP). MAP preferentially invades M cells by creating a fibronectin bridge and causes subsequent invasion of subepithelial macrophages. Entry into the macrophage is accomplished by ManLAM binding to CR3 and mannose receptors. MAP invasion into the lamina propria may also be gained through intestinal epithelial cells by an unknown mechanism. Dendritic cells may also transport MAP inside the lamina propria during sampling through tight junctions. MAP interaction with the dendritic cell receptor, DC-SIGN, may prime and promote a Th2 response. TLR, Toll-like receptor.
due to the lack of lysosomes and hydrolytic enzymes present in these cells (Miller et al., 2007). Therefore, many antigenic properties of MAP would remain unaltered after passing through M cells. It is well established that fibronectin (FN) attachment proteins present on MAP facilitate FN binding of the bacterium, which in turn forms a FN bridge with β1 integrins located on intestinal epithelial cells (SigurSardottir et al., 1999; Pieters, 2001; Secott et al., 2001, 2004).
More recent studies have shown that passing through epithelial cells alters MAP’s characteristics and changes the response of macrophages (Everman et al., 2015). Preferential binding of MAP to M cells may be explained by the high density of β1 integrin present on the luminal surface of these cells in comparison with other cell types, such as enterocytes. Villous epithelial cell invasion is due to an unknown FN-independent mechanism (Secott et al., 2001, 2004). However, MAP preference for M cells appears to require more than just FN-integrin interactions, since the closely related M. avium subsp. avium enters the intestinal wall via absorptive epithelial cells, despite presence of FN attachment protein genes (Secott et al., 2002).MAP primarily resides within macrophage cells; however, some studies suggest that epithelial cell processing prior to macrophage exposure may enhance efficiency of invasion into macrophages. MAP exposed to Mac-T cells, a mammary epithelial cell line, displayed increased invasion efficiency in subsequent infections of MDBK cells. Increased invasion efficiency by prior exposure may be due to upregulation of MAP3464, encoding an oxidoreductase that activates host Cdc42 and Rho internalization pathways (Patel et al., 2006; Alonso-Hearn et al., 2008). DNA microarray analysis of MAP 24h post-infection of Mac-T cells revealed upregulation of 20 MAP genes related to regulatory, metabolic and virulence-associated functions compared with MAP grown in Middlebrook 7H9 broth cultures. From this gene set, Patel et al. (2006) hypothesized that a 35-kDa MAP protein (major membrane protein, or MMP), which previously was shown to enhance invasion in epithelial cells, may be upregulated in response to prior exposure to Mac-T cells (Bannantine et al., 2003). Following up, Abdellrazeq et al. (2018) observed cytotoxic T lymphocyte (CTL) responses to a MAP ∆relA strain and found this response targeted MMP. This CTL-mediated response was recapitulated by stimulating bovine dendritic cells ex vivo with MMP alone, showing killing of intracellular MAP and signifying the importance of MMP in infection.
This group has more recently published a nanoparticle-based peptide vaccine incorporating MMP to provoke CD8 T-cell-mediated MAP killing (Abdellrazeq et al., 2019).Following M-cell invasion into the sub- epithelial dome, MAP may encounter dendritic cells and/or macrophages. It is well established that MAP interacts with intestinal dendritic cells through its cell wall glycolipid mannosylated lipoarabinomannan (ManLAM) and the dendritic cell receptor DC-SIGN (Jozefowski et al., 2008). MAP may use intestinal dendritic cell invasion as a strategic manoeuvre, since the primary function of intestinal dendritic cells is to sample and present commensal bacteria through tight junctions to the gut-associated lymphoid tissue. Thus, MAP would be able to overcome tight junction barriers and be directly transported to the lamina propria to interact with subepithelial macrophages. Furthermore, ManLAM-DC-SIGN interaction may prime MAP to direct a Th2 response, which would lead to immune subversion, as suggested for M. tuberculosis (Jozefowski et al., 2008).
Although murine monocyte-derived dendritic cells (MoDCs) are capable of phagocytizing MAP and secrete IL-10 after doing so (Basler et al., 2013), not much is known about their interaction. Inside dendritic cells (DCs), M. tuberculosis vacuoles cannot access exogenous material or biosynthetic pathways and fail to replicate (Tailleux et al., 2003), which is also seen with Mycobacterium avium (Salte et al., 2011). Like macrophages however, the vacuole is arrested and does not acidify (Tailleux et al., 2003). MAP- infected DCs also fail to mature properly when cultured in media previously used for MAP/ MDM culture (Basler et al., 2013). DC's inability to mature in the presence of MAP was also observed in tissues (Lei et al., 2008).
As with dendritic cells, ManLAM from MAP is capable of interacting with macrophage cell surface receptors. The best-documented interaction is between MAP and the mannose receptor, which enhances macrophage phagocytosis of MAP (Pieters, 2001; Gatfield and Pieters, 2003; Rowe and Grant, 2006; Souza et al., 2007a).
Upon entry into macrophage cells, there is simultaneous replication of MAP and bacterial killing by the host, spurring on an initial Th1-like response (Rowe and Grant, 2006; Alonso et al., 2007; Woo et al., 2007). Initial killing of MAP may be due to a rapid phagosome acidification response from the host, allowing phagosome-lysosome fusion to occur in some cells. The end result of phagosome-lysosome fusion would presumably be destruction of MAP and presentation of antigens to T cells via major histocompatibility complex (MHC). However, most phagosomes containing MAP and other pathogenic mycobacteria fail to mature. Studies of interactions between M. tuberculosis ManLAM and macrophages indicate that ManLAM is indispensable for blockage of phagosome maturation (Russell et al., 2002; Yates and Russell, 2005). The ability of MAP and other mycobacteria to inhibit phagosome-lysosome fusion is essential to prevent pathogen awareness by the host immune system, thus allowing MAP to hijack macrophage resources and persist unabated. An active role for MAP in preventing phagosome-lysosome fusion is supported by the observation that live MAP is able to persist within phagosomes for 15 days, while phagosome function is not interrupted following uptake of killed MAP (Kuehnel et al., 2001). Other macrophage receptors that are important for MAP binding include those for complement, immunoglobulin, transferrin, scavengers and surfactant protein A (Pieters, 2001; Souza et al., 2007a). MAP binding to one complement receptor prevents activation of an oxygen burst and is abrogated with addition of monoclonal antibodies (SigurSardottir et al., 2004).The efficiency with which MAP enters macrophage cells appears to differ with respect to MAP genotype, such that species-specific variation is observed (Gollnick et al., 2007). Despite variation in invasion efficiency, MAP strains all seem to employ a similar infection protocol or modus operandi. Zhu et al. (2008) determined that expression patterns from three MAP strains of different types based on short sequence repeats showed upregulation of 2 7, 22 and 35 genes, respectively, when isolated from infected bovine monocyte-derived macrophages (MDMs) at 48 and 120h post-infection.
Many of the genes on these lists were similar or had overlapping cellular functions. Pathway analysis categorized gene functions related to small-molecule degradation, energy metabolism, amino acid biosynthesis, lipid biosynthesis, broad regulatory functions, synthesis and modification of macromolecules, cell envelope, transport/binding proteins, virulence, antibiotic production and resistance, and conserved hypothetical proteins. All three MAP strains upregulated MAP4041c and MAP4281, which are suggested to play a role in protein transport and act as insertion elements, as well as genes related to lipid degradation, membrane transportation and DNA repair at 48 h. Taken together, this comparative transcriptional analysis suggests that diverse MAP genotypes use a similar modus operandi for survival in the host.Previous studies have investigated the ability of MAP strains to regulate expression of MHC molecules during macrophage infection. Although MAP infection of J774 murine macrophages did not affect expression of MHC class II molecules, antigen presentation decreased, which may be due to MAP limitation of antigen processing (Kuehnel et al., 2001). However, these results conflict with those demonstrating downregulation of MHC class I and class II molecules in bovine macrophages infected with live and killed MAP (Weiss etal., 2001). It is exciting to speculate on the potential that one or a number of hypothetical genes identified by Zhu et al. (2008) may be responsible for MAP regulation of MHC class I and class II molecules and control of antigen processing.
While immediate activation of MAPK signalling by MAP is of obvious importance in the response of macrophages to infection, studies focused on this cannot tell us what effect MAP might be having on the ability of macrophages to respond to T cells and to activate these cells to respond to infection. One of the most critical components of macrophage-T-cell interactions is engagement of CD40 on macrophages by CD154 (CD40 ligand) on activated T cells.
CD40-CD154 binding is one of the major mechanisms leading to macrophage activation via T-cell interactions, and soluble CD154 can mimic many of the processes observed when T cells activate macrophages (Grewal and Flavell, 1996, Grewal and Flavell, 1998; Grewal et al., 199 7). CD40 is a member of the TNF receptor superfamily and is expressed on numerous cell types, including B cells, mono- cytes/macrophages, endothelial cells, dendritic cells, fibroblasts and vascular smooth muscle cells (Clark et al., 1996; van Kooten and Banchereau, 1996). Accordingly, typically over 70% of bovine MDM stain positive for cell surface CD40 after 7 days of maturation in culture (Chiang et al., 2007). In monocytes and macrophages, CD40 signalling leads to secretion of inflammatory cytokines including IL- 12, chemokines including β-chemokines (Stout and Suttles, 1996; di Marzio et al., 2000) and matrix metalloproteinases (Malik et al., 1996); induction of inducible nitric oxide synthase (INOS); production of nitric oxide (Tian et al., 1995; van Kooten and Banchereau, 1996); enhanced cell survival; and induction of costimulatory molecules (Kiener et al., 1995). T cells derived from CD154-deficient mice are impaired in their ability to induce macrophage effector functions (Stout and Suttles, 1996), and consequently these mice are highly susceptible to intracellular pathogens that would otherwise have been cleared by an appropriate T-cell-macrophage interaction (Soong et al., 1996).Studies on CD40 signal transduction have resulted in a complex picture of different mediators and pathways involved. Two major signalling pathways are activated downstream of CD40, which both involve activation of latent transcription factors. One pathway involves activation of the inhibitor of nuclear factor kappa B kinase complex, leading to nuclear translocation of active nuclear factor kappa B. The second mechanism is activation of the MAPK pathway, a cascade of phosphorylation events that primarily results in post-transcriptional activation of transcription factors like cyclic adenosine monophosphate (cAMP)-response element binding protein, activating transcription factor, Ets, and AP-1 (van Kooten and Banchereau, 1996, 2000). Both pathways synergize in inflammatory gene expression, including expression of IL-12p40, iNOS, IL-6, IL-8 and TNFα. Not surprisingly, CD40-CD154 signalling is a target for many intracellular pathogens. For example, Mathur et al. (2004) demonstrated that Leishmania major, an intracellular parasite causing leishmaniasis in humans, is able to inhibit CD154-CD40-mediated IL-12p40 and iNOS gene expression in murine peritoneal macrophages. This blockade appears to involve interference with activation of two main members of the MAPK pathway, p38 and ERK1/2 (Awasthi et al., 2003; Mathur et al., 2004).
MAP-infected macrophages are defective in some aspects of CD40 signalling (Sommer et al., 2009). In uninfected macrophages, CD40-CD154 binding results in large increases in TNFα, IL-6, IL-10, IL-8, IL-12p40 and iNOS gene expression within 6 h. In MAP- infected macrophages, TNFα and IL6 gene expression following CD40-CD154 binding is relatively unaffected. In contrast, MAP- infected macrophages fail to activate expression of IL-12p40 and iNOS gene expression. This is a critical difference, since IL-12 is a major driving force for development of an appropriate Th1-like response and production of IFNγ by T cells. In macrophages, iNOS activity and production of reactive nitrogen species is a major mechanism used to kill phagocytosed bacteria. For an intracellular bacterium such as MAP, limiting production of IL-12 and iNOS would ensure survival and development of an inappropriate immune response, particularly in the face of enhanced IL-10 production. It has also been suggested that failure to properly activate and/or engage T cells could lead to development of regulatory T cells, which would further reduce Th1-like immune activity against MAP (de Almeida et al., 2008). Results of Sommer et al. (2009) are also consistent with observations in vivo (Coussens et al., 2004), where MAP-infected intestinal tissues contained elevated levels of IL-10 but not IL-12p40 or IL-12p35. These tissues also contain elevated levels of TGFβ (Coussens et al., 2004; Khalifeh and Stabel, 2004). TGFβ can be produced by regulatory Th3 cells and by regulatory γδ T cells, among others. These defects may be more important in early responses to MAP infection however, since despite the presence of IL-10, T regulatory cells seem to be largely absent in later stage MAP-infected tissues (Roussey et al., 2016).
9.4
More on the topic Secret Operations: Intestinal Epithelial Cells, Macrophages and MAP:
- HIV is a virus that infects white blood cells, primarily those called CD4 cells (also called T4 cells or T-helper cells).
- SOLUBLE FACTORS FROM MACROPHAGES/MICROGLIA EXPOSED TO HIV-1
- Epithelial Migration
- Acidophilic Macrophage Pneumonia/ Epithelial Hyalinosis
- Failure of Epithelial Migration
- Innate Response to MAP Infection
- 11 Failure of Epithelial Migration: Cerumino liths
- Fabrication of Nationalist Plots by the Secret Police in Ukraine, 1929-341
- Intestinal Plasmacytosis
- “The Beginning of the War Will Be Secret” — Jenny Holzer, “Survival,” 1983[CXX] [CXXI]
- 24 Small intestinal diarrhoea
- Comparison of the Virulence and Pathogenicity of MAP Strains
- Intestinal Pseudo-Obstruction (Ileus)
- Eimeria spp. Infection: Intestinal Coccidiosis
- ACUTE INTESTINAL OBSTRUCTION
- Intestinal Obstruction and Rupture
- HIV KILLING OF INFECTED T CELLS