VIRAL LIFE CYCLE
Lentiviruses have a characteristic conical core structure that distinguishes them from type C retroviruses. In addition to Gag, Pol, and Env, three to six additional proteins with diverse regulatory roles contribute to their genetic complexity.
HIV-1 encodes six such proteins: Tat, Rev, Nef, Vif, Vpr, and Vpu.Early Events
The main molecular events of HIV-1 (Figure 1.1) entry are now well understood, although the role of the viral envelope protein in cell death continues to be enigmatic.59,60 The initial event in virus entry is the binding of the processed viral envelope glycoprotein (gp120) to the CD4 molecule on the cell surface. However, for years, it was known that introduction of human CD4 into nonhuman cells was insufficient to confer entry. The implicated second receptor was determined to be a group of chemokine co-receptors in 1996—CCR5 and CXCR4 predominated.61 Subsequently, great structural detail has been revealed. After CD4 binding, a conformational shift seems to occur within gp120, which exposes a chemokine co-receptor binding site.62 The dominant co-receptors used by HIV-1 are CXCR4 and CCR5. CXCR4-using (or X4) viruses efficiently infect T cells and are also referred to as T cell tropic. CCR5 (or R5) viruses efficiently infect cells of the monocyte/macrophage lineage and are referred to as macrophage tropic.
The limited exposure of the key surfaces within the critical intermediate structures that occur during gp120 binding and fusion is a main reason for the failure to generate neutralizing antibodies
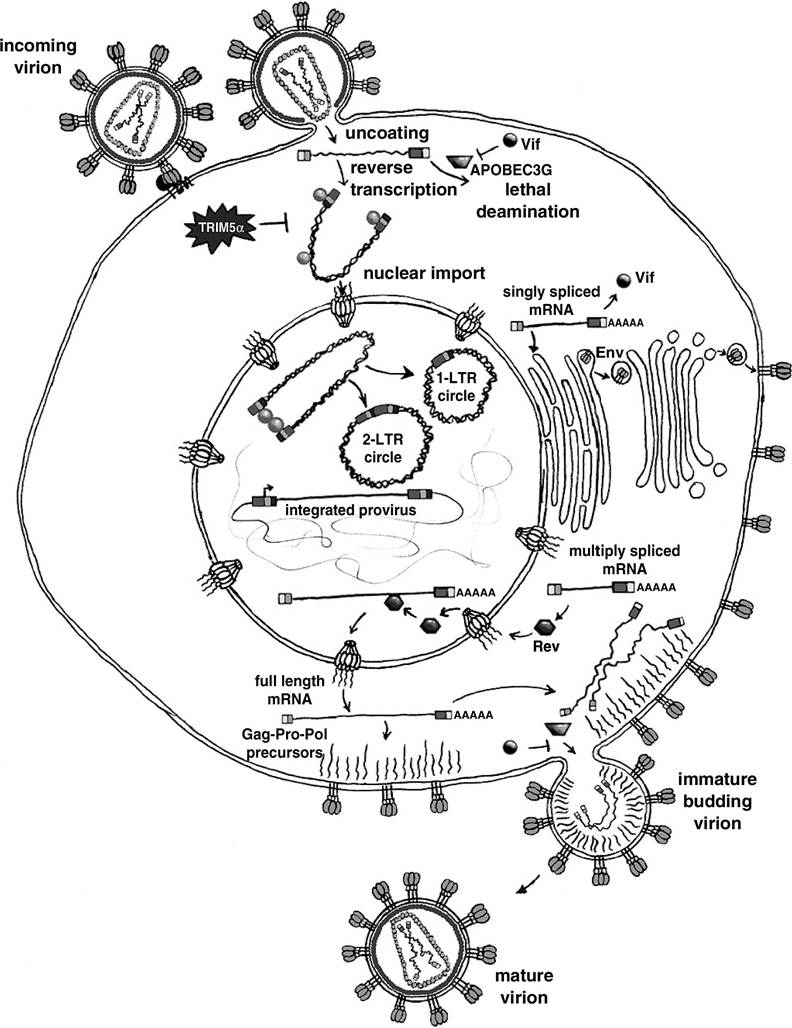
FIGURE 1.1 The HIV-1 life cycle. Entry is illustrated in a temporal sequence beginning at upper left, with assembly and budding at the lower right. The pre-integration complex (PIC) is depicted as a U-shaped intermediate with integrase protein molecules, presumably multimers, at the termini.
TRIM5alpha81 acts in the target cell via interaction with the capsid protein. In contrast, APOBEC3G is incorporated in the producer cell and subsequently appears to cause deamination of the minus strand DNA at C residues, leading to G to A hypermutation. Viral protease-mediated maturation of the core is synchronous with budding. (Drawing by Dyana T. Saenz.)capable of controlling the infection.63 Binding to the co-receptor induces further conformational changes within gp120 that dislocate it from gp41 and expose the gp41 fusogenic domain,64 an amino-terminal hydrophobic peptide that inserts into the cell membrane. The gp41 then collapses into a trimer-of-hairpins structure that brings the two membranes close and mediates fusion.65 Chemokine co-receptor usage seems to be fundamental to lentiviral biology, because FIV also uses CXCR4.66'67 The recent identification of feline CD13468 as the primary receptor rather than feline CD4 (or CD969) explains the additional tropism of FIV for B cells and CD8 cells. Humans with a homozygous mutation of the CCR5 gene that prevents the receptor from being presented at the cell surface are less likely to be infected by HIV, and if infected, they progress to AIDS less rapidly.70-72
Cell-cell fusion mediated by gp120 (syncytium formation) and cell surface signal transduction triggered by gp120 can be shown to cause cell death in vitro; their roles in vivo remain unclear.73 It is clear, however, that inhibition of entry can be used therapeutically. T-20 (enfurtivide) is an FDA-approved synthetic peptide that targets the amino-terminal region of gp41, which becomes exposed in the transient extended state.74 This drug must be combined with other antiviral agents targeted at different stages of the life cycle or resistance develops rapidly.
Uncoating and Transport to the Nucleus
The uncoating, trafficking, and maturation steps that follow the entry of the viral core into the target cell cytoplasm constitute a poorly understood interval in the HIV-1 life cycle.
HIV-1 seems to hijack a microtubule-dependent motor, dynein, to travel to the vicinity of the nucleus.75 In this respect, it resembles other viruses.76-79 However, we know little regarding the details of HIV-1 uncoating and the structure of the preintegration complex (PIC) as it evolves during trafficking. During this phase, there is a complex interplay between cellular restriction factors80-82 and cellular factors that are exploited by the virus.83 Recently, a viral defense mechanism that protects the integrity of the genome against a cellular antiretroviral activity was identified.84 The virally encoded Vif (virion infectivity factor) protein, long known to enhance infectivity in a manner that was intriguingly cell-type dependent and to display a phenotype dependent on the producer cell rather than on the target cell, has been shown to promote degradation of the cellular protein APOBEC3G by targeting it to the proteasome.85-91 In the absence of Vif, APOBEC3G, a cytidine deaminase, is incorporated into virion particles in the producer cell. APOBEC3G then mediates deamination of deoxycytidine to deoxyuridine within the minus-strand cDNA in the target cell, thus resulting in lethal G to A hypermutation of the viral genome.Controversy persists about the underlying mechanisms by which lentiviral PICs transit the nucleopore complexes of mitotically inactive cells.92 Proposed effectors have included peptide determinants in matrix,93-100 Vpr,96,101,102 and integrase.98,103 The central DNA flap104-106 has also been implicated.104,105 Importin-7 has been suggested to mediate PIC translocation in semipermeabilized cell assays.107 Countervailing views or reassessments exist for some of these proposals.92,108-111,112 (See references111,112 for reviews.)
Integration
The determinants of subsequent intranuclear preintegration PIC trafficking are not understood at all.
However, its outcome is now known to be a biased distribution of HIV-1 integration sites, with actively transcribed genes being favored.113,114 How cellular factors govern this important selectivity is unknown. In addition, little is known regarding how cellular proteins participate in the HIV-1 integration reaction as it occurs in cells.115The linear viral cDNA that results from reverse transcription has four principal fates: bona fide integration; formation of 1-LTR circles, probably by homologous recombination116; conversion to 2-LTR circles by the host cell nonhomologous DNA end-joining (NHEJ) pathway117; and autointegration to form defective DNAs.118 Only the linear form is a precursor to integration. Details of integrase catalysis have been reviewed elsewhere.119
There is some evidence that integration intermediates might trigger cell death under some circumstances (e.g., when DNA repair mechanisms are impaired). Early work by Temin and colleagues suggested that transient cytopathic effects correlated temporally with the transient accumulation of large numbers of unintegrated spleen necrosis virus DNA molecules.120,121 More recently, the nonhomologous end joining (NHEJ) pathway has been implicated in completing the final stages of integration, and these studies have raised the possibility that integration intermediates can be toxic.122 The NHEJ pathway is the principal repair system for double-stranded DNA breaks in eukaryotic cells. Such breaks occur during lymphocyte V(D)J recombination. They are also generated by ionizing radiation and are proapoptotic: just a few double-strand DNA breaks can induce p53-dependent G1 arrest.123 The principal components of this pathway are DNA-dependent protein kinase (DNA-PK), Ku, XRCC4 ligase, and DNA ligase IV.124,125 DNA-PK, which is activated by dsDNA ends, participates in this repair along with the Ku proteins, although the exact role played by DNA-PK has not been established.
This pathway is currently understood to repair doublestrand gaps and not single-strand gaps, which is of relevance to the following discussion.125 Severe combined immunodeficient (SCID) mice have a truncation mutation in both alleles of DNA-PKCS, so they carry out the initial double-strand cleavage steps of V(D)J recombination but cannot repair the resulting double-strand breaks.126 This defect results in an absence of mature B and T cells and greater susceptibility to spontaneous malignancy.Retroviral reverse transcription also generates a double-stranded DNA with blunt ends. Before integration, two nucleotides are removed by integrase from each 3' strand of the linear molecule. The recessed 3' ends are then joined (the strand transfer reaction) to 5' DNA ends in cellular DNA via a direct attack by the 3' hydroxyl group.119
Daniel et al. found that retroviral vector integration was reduced in SCID cells and that high- titer infection induced apoptosis within approximately 12 h of infection.122 In contrast, integrationdefective vectors did not trigger cell death. They concluded that DNA-PK is required to complete integration, presumably by repairing the DNA gaps. Subsequent reports have led to refinement and some contradiction of this scenario.117,127,128 In one study, increased cell death was also observed in SCID cells, but only after transduction at quite high multiplicity of infection (MOI) and subsequent cell division.127 At an MOI of less than 1.0, transduction efficiency was actually higher in SCID cells than in control cells, and it proceeded normally in vivo in SCID mice brain without cell death, leading to the conclusion that DNA-PK and another DNA repair enzyme, PARP, are not essential for HIV-1 integration.127 Similarly, Li et al. found that lower-titer HIV-1 vector infection proceeded normally in cells mutant for components of the NHEJ pathway.117 Similar to other studies, high-titer infections were found to be toxic in NHEJ-mutant cells, apparently by an apoptotic mechanism.
However, a key difference is that integrase mutant viruses were just as toxic, suggesting that it is unintegrated viral DNA ends that act as the apoptotic signal rather than unrepaired viral-cellular junctions. Unintegrated DNA was also found to be a substrate for the NHEJ pathway in that it was required for 2-LTR circle formation.117The roles of DNA-PK and other cellular repair mechanisms in integration and cell death remain uncertain. Its relevance to the actual disease pathology of lentiviruses is not clear, because normal human CD4+ T cells are fully competent for the NHEJ pathway.
Latency
The factors that influence PIC trafficking to particular integration sites after nuclear import are unknown, but they are of clinical importance since recent data established that HIV-1 integration sites are not randomly distributed in the genome. This, in turn, may influence the ability of latent proviruses to serve as sources of recrudescence from resting memory T cells. Postintegration proviral latency is an area of intense interest at present. Viral rebound invariably occurs after the discontinuation of HAART, generally within 2 weeks, and virus can be isolated from T cells in vitro during effective HAART'.129131 The capacity to remain transcriptionally silent for extended durations is one factor that permits the virus to evade immune surveillance.132 The half-life of the latent reservoir in resting memory T cells is estimated to be approximately 4 years, indicating that eradication by decay on HAART is highly unlikely to be feasible.133,134 Therefore, understanding the molecular mechanisms controlling integration site preference, silencing, and reactivation of the integrated HIV-1 provirus has important implications for understanding HIV disease pathogenesis and for therapy.134,135 Chromatin possesses marked structural and functional heterogeneity, and extensive evidence demonstrates that retroviral transcription is strongly affected by the context of integration.136-139 Epigenetic mechanisms such as histone deacetylation and DNA methylation play major roles in the silencing of viral promoters that occur progressively after integration.140,141 Methylation-associated silencing is dependent on chromosomal position.140
Recent studies established definitively that several retroviruses do not integrate randomly with respect to genomic loci. MLV and HIV-1 both integrate preferentially into transcriptionally active regions.113,114,142, 143 However, the placements of their proviruses seem to differ with respect to functional elements within the cellular transcription units. HIV-1 integration favors regions downstream of promoters.113,114,142 In contrast, MoMLV integrations show a predilection for promoter regions, favoring the 2.5 kb surrounding the transcriptional start site.114 In latently infected cells, a bias toward integration in heterochromatic regions, such as alphoid repeats, is also observed.144 Integration site distributions will receive concentrated study with the newer methods available113,114,142 because of the implications for HIV-1 latency134,136,144-146 and to address the problem of insertional mutagenesis in human gene therapy.147
Cellular cofactors responsible for retroviral PIC trafficking or integration site specificity have not been identified.148 Lentiviral integrase proteins localize to cell nuclei and associate with chromatin when expressed in the absence of other viral components.98,149-153 These observations have led to efforts to identify a nuclear localization signal (NLS) and to implicate this protein in preintegration complex nuclear translocation. However, nuclear/chromatin targeting of lentiviral integrases now seems to be due not to any nuclear localization signal but rather to interaction with the transcriptional coactivator LEDGF∕p75.153,154 This interaction, which is lentivirus specific, is clearly not needed for nuclear import of the PIC,153 but there is interest in its potential involvement in integration site choice. Interactions of integrase or PIC DNA with other cellular factors have been demonstrated as well.148,155,156 Examples are BAF,146,157,158 HMGa1 protein,159-162 INI1,155,163 Ku70 and Ku80,117 the aforementioned DNA-PK,122 and hRad18.164 Some are known to enhance in vitro integration reactions,155,157,165-169 as does the viral nucleocapsid (NC) protein,159,169 although the significance in each case to the reaction in the natural life cycle of HIV-1 is not clear. INI1, an essential constituent of the human SWI/SNF chromatin remodeling machine, is incorporated into HIV-1 but not HIV-2, SIV, or other retroviral particles170 and it seems to be required for efficient HIV-1 budding.163 INH also stimulates the DNA-joining activity of HIV-1 integrase in vitro, and a role in targeting the viral DNA to active genes has been hypothesized.155 Moreover, incoming HIV-1 and murine leukemia virus (MLV) PICs were found to trigger export of INI1 and the nuclear body constituent PML (promyelocytye leukemia protein) to the cytoplasm, which may be part of a cellular antiviral response.168
Late Events
Two small RNA-binding regulatory proteins, Tat and Rev, govern viral mRNA production. HIV-1 transcription is minimal in the absence of Tat, because transcriptional elongation does not occur efficiently. Tat binds to an RNA stem-loop (TAR) in the 5 end of the nascent viral transcript. The N-terminal domain of Tat interacts with CyclinT1, which, in turn, recruits the cyclin-dependent kinase 9 (CDK-9), leading to hyperphosphorylation of the large subunit of the RNA polymerase II.171,172 The result is increased elongation efficiency of the Pol II complex.172,173 Tat’s role in modulating transcription seems to extend to the cellular Iranscriptome174-176; roles in regulating apoptosis,177 and class I major histocompatibility complex (MHC) molecule expression have also been reported.178 Rev binds to a secondary RNA structure in the env region, the Rev response element, and enables the virus to solve a central problem: export of its unspliced genomic RNA against the normal cellular checkpoint that prevents export of intron-containing messages.179,180 The Nef and Vpr proteins have been reported to play a wide variety of roles in the viral life cycle and to be implicated in cell death; these proteins will be reviewed in Chapters 15 and 14, respectively.
Maturation of the virion via protease cleavage is synchronous with budding. Recently, viral budding has been shown to employ a complex cellular machinery involved in protein sorting and membrane budding at the multivesicular body181; this system is involved in the intracellular budding of HIV-1 virions that is observed in macrophages.182,183 The HIV-1 protease cleaves the Gag/Pol precursor into virion proteins and was also implicated in cleaving cellular proteins such as caspase- 8,184 a cell death mechanism that will be reviewed in Chapter 17.
More on the topic VIRAL LIFE CYCLE:
- VIRAL PROTEIN R (VPR): STRUCTURE AND FUNCTION IN THE VIRAL LIFE CYCLE
- Life tables can be based on age, size, or life cycle stage
- Gender, Identity, and Life-Cycle Rituals
- CASH FLOW AND THE COMPANY LIFE CYCLE
- Life-Cycle Rituals
- Life-Cycle Events
- Life Cycle Rituals
- LIFE CYCLE STRATEGIES AMONG PROTOZOA
- PROTEASE (PR) IN VIRUS LIFE CYCLE AND AIDS PATHOGENESIS
- Life Cycle Evolution